98) Plummer-Vinson Syndrome :
(Paterson-Kelly syndrome, Hysterical dysphagia, Sideropenic dysphagia)
Etiology :
Iron deficiency is the primary cause of the syndrome. Chronic blood loss (profuse menstruation), inadequate dietary intake, faulty iron absorption or increased requirements of iron can lead to iron deficiency.
Clinical features :
Plummer-Vinson syndrome occurs chiefly in women in 4th to 5th decades of life.
a) Oral manifestations :
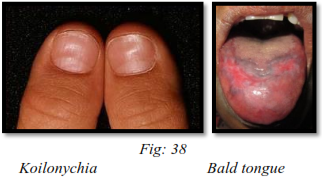
Cracks or fissures at the corner of the mouth: the vermillion border of lip is thinned and mouth is narrowed. The tongue is smooth, devoid of papillae, and usually red in color. It is often painful and edematous. Leukoplakia of the oral mucosa has been reported in a number of cases, especially on the dorsum of tongue.
General appearance :
The facial skin is usually pale, dry, smooth and atrophic, giving the patient a characteristic asthenic appearance. The nails are brittle and spoon shaped (koilonychia).
c) Esophagus :
Dysphagia is the outstanding feature. The difficulty in swallowing is attributed to formation of esophageal stricture or web; which occurs at third or sixth cervical vertebrae. The web is described as a thin, crescentic membrane arising usually from the anterior esophageal wall. The mucous membranes of oral cavity and esophagus are atrophic and show loss of normal keratinization. The atrophy of mucous membranes of upper alimentary tract predisposes to the development of carcinoma (therefore it is a pre cancerous condition).
99) Popliteal Web Syndromes:
Popliteal pterygium syndrome (PPS) is an inherited condition affecting the face, limbs, and genitalia. The syndrome goes by a number of names including the popliteal web syndrome and, more inclusively, the facio-genito-popliteal syndrome. The term PPS was coined by Gorlin et al.. in 1968 on the basis of the most unusual anomaly, the popliteal pterygium (a web behind the knee).
Clinical features
The genetic locus for PPS was localized to chromosome 1. The disorder is inherited in an autosomal dominant manner and is due to mutation of the IRF6 gene. Most reported cases are sporadic; advanced parental age is found in a number of these cases, suggesting new mutations.
The term PPS has also been used for two rare autosomal recessively inherited conditions: Lethal PPS and PPS with Ectodermal Dysplasia. Although both conditions feature a cleft lip/palate, syngnathia, and popliteal pterygium, they are clinically distinguishable from the autosomal dominant case. Lethal PPS is differentiated by microcephaly, corneal aplasia, ectropion, bony fusions, hypoplastic nose and absent thumbs, while PPS with Ectodermal Dysplasia is differentiated by woolly hair, brittle nails, ectodermal anomalies, and fissure of the sacral vertebrae.
100) Prader-Willi Syndrome :
It is a rare genetic disorder in which seven genes (or some subset thereof) on chromosome 15 (q 11–13) are deleted or unexpressed (chromosome 15q partial deletion) on the paternal chromosome.
The variety of symptoms can range from poor muscle tone during infancy to behavioral problems in early childhood. Some symptoms that are usually found in infants, besides poor muscle tone, would be a lack of eye coordination, some are born with almond-shaped eyes, and due to poor muscle tone the infant may fail to have a strong sucking reflex. Their cry is weak, as well as the difficulty of waking up. Another sign of this disease is a thin upper lip.
Oral features :
1. Dental caries
2. Enamel hypoplasia
3. Malocclusion
4. Heavy calculus
5. Decreased salivation
6. Viscous, bubbly saliva
7. Gingivitis
8. Fish-like mouth with triangular shaped upper lip
9. Microdontia
10. Thick saliva at edges of mouth
11. Arched palate
101) Proteus syndrome:
(Wiedemann syndrome)
It is a congenital disorder that causes skin overgrowth and atypical bone development, often accompanied by tumors over half the body. Proteus syndrome is a progressive condition wherein children are usually born without any obvious deformities. Tumors of skin and bone growths appear as they age. The severity and locations of these various asymmetrical growths vary greatly but typically the skull, one or more limbs, and soles of the feet will be affected. There is a risk of premature death in affected individuals due to deep vein thrombosis and pulmonary embolism caused by the vessel malformations that are associated with this disorder.
Clinical features:
102) Pterygopalatine Fossa Syndrome :
This syndrome resembles Trotter’s syndrome. It is due to tumor metastases to pterylgopalatine fossa. (The fossa contains maxillary division of trigeminal nerve, internal maxillary artery and sphenopalatine ganglion).
Clinical features :
Include pain in upper teeth, anesthesia of infraorbital region, blindness, anesthesia of palate and motor nerve paralysis of pterygoids.
103) Raeder’s syndrome :
(Paratrigeminal Syndrome)
History for Raeder paratrigeminal syndrome suggests trigeminal nerve involvement with pain, sensory or motor deficits, and/or ipsilateral oculosympathetic paresis. It resembles Horner syndrome and manifests as oculosympathetic paresis with ptosis and miosis. Unlike Horner syndrome, facial sweating and ipsilateral trigeminal sensory irritation are preserved, leading to production of facial pain. Other parasellar cranial nerves also may be involved and an enophthalmos also may be apparent. The pain associated with Raeder syndrome is deep and boring and is localized in or around the eye. Intermittent, lancinating pain also may occur. Typically self-limited, the pain usually remits in 2-3 months.
The pain occasionally follows a recurrent pattern. It can be associated with conjunctival tearing, erythema, enophthalmos, and decreased intraocular pressure.
Clinical features :
1. Headache or pain in trigeminal nerve distribution.
2. Ocular symphathetic paralysis.
104) Ramsay hunt syndrome:
(Herpes Zoster Oticus)
It is a disorder that is caused by the reactivation of pre-existing herpes zoster virus in the geniculate ganglion, a nerve cell bundle, of the facial nerve.
Ramsay Hunt syndrome type 2 typically presents with inability to move many facial muscles, pain in the ear, taste loss on the front of the tongue, dry ears and mouth, and the eruption of an erythematous rash.
Clinical features :
1. Zoster inection of Geniculate ganglion with involvement of external ear and oral mucosa.
2. Facial paralysis, pain of external auditory meatus and pinna of the ear.
3. Vesicular eruption in the oral cavity, and oropharynx with hoarseness, tinnitus and vertigo.
105) Reiter’s Syndrome :
Etiology :
The etiology is still unclear. There is evidence of infectious origin. In recent years pleuro pneumonia like organisms (PPLO) have been implicated and even more recently, a Bedsonia group of virus has been isolated from the patients. An abnormal immune response to microbial antigens is regarded as a likely mechanism for multiple manifestations of this syndrome.
Clinical features :
It most commonly occurs in men between the age group of 20 and 30 years. Above the age of 50, the disease is seldom seen. There is typical tetrad of manifestations, which include urethritis, arthritis, conjunctivitis and mucocuta neous lesions. The onset of symptoms may be preceded by weight loss, fatigability and diarrhea. The urethritis generally precedes the appearance of other lesions. The urethral involvement consists of discharge associated with itching and burning sensation. The examination of discharge reveals no bacteria.
Oral manifestations :
The oral lesions are described as aphthous type ulcerations by Pindborg which are usually painless, red, slightly elevated areas, sometimes granular or even vesicular, with a white circinate border. The lesions can occur anywhere in oral cavity. The tongue may exhibit superficial erosions similar to that of geographic tongue.
106) Rieger’s Syndrome :
(Hypodontia, Mesoectodermal dysgenesis of the iris & cornea and myotonic dystrophy)
It is transmitted as an autosomal dominant trait.
a) Oral manifestations :
b) Face :
The face appears wide and there is prognathic mandible. This can just be a relative prognathism due to under development of maxilla or due to loss of vertical height due to hypodontia.
107) Riley-Day Syndrome :
(Familial dysautonomia, Familial autonomic disorder)
It is a hereditary sensory and autonomic neuropathy type III (HSAN-III) — is a disorder of the autonomic nervous system which affects the development and survival of sensory, sympathetic and some parasympathetic neurons in the autonomic and sensory nervous system resulting in variable symptoms including: insensitivity to pain, inability to produce tears, poor growth, and labile blood pressure Etiology:
Familial dysautonomia is the result of mutations in IKBKAP gene on chromosome 9, which encodes for the IKAP protein (IkB kinase complex associated protein) and is inherited in an autosomal recessive fashion. Both parents must be carriers in order for a child to be affected
Clinical features :
1. There are a variety of autonomic, motor and sensory dysfunctions which result in vasomotor problems. Reflexes are absent.
2. Absence of tears, postural hypotension, excessive perspiration,
3. Feeding difficulties, disturbed swallowing reflex, and speech difficulties.
4. Coldness of extremities.
5. Generalized retardation of growth and
6. Scoliosis has been reported.
Oral manifestations :
1. Drooling of saliva due to disturbed swallowing reflex and diminished pain and taste sensation have been reported.
2. Fungiform and circumvallate papillae can be congenitally missing or rudimentary while filiform papillae are not affected.
108) Rutherford’s Syndrome :
The syndrome was first described by Rutherford in 1931. It is transmitted as an autosomal dominant trait.
Clinical features :
1. Clinical features include mild gingival fibromatosis,
2. Dense curtain-like corneal opacities involving the superior part of each cornea.
3. Failure of eruption of teeth.
4. The onset and degree of gingival fibromatosis are unrelated to the eruption of the dentition into the oral cavity. The tooth eruption can be delayed or only few teeth may erupt or the complete complement of both dentitions may remain within the alveolus, covered by dense fibrotic gingiva.
5. Patient can exhibit aggressive behavior and mental retardation.
109) Rubinstein-Taybi syndrome :
(Broad Thumb-Hallux Syndrome , Rubinstein Syndrome)
It is characterized by short stature, moderate to severe learning difficulties, distinctive facial features, and broad thumbs and first toes. Other features of the disorder vary among affected individuals. People with this condition have an increased risk of developing noncancerous and cancerous tumors, leukemia, and lymphoma. This condition is sometimes inherited as an autosomal dominant pattern and is uncommon
Etiology:
Mutations in the CREBBP gene cause Rubinstein–Taybi syndrome. The CREBBP gene makes a protein that helps control the activity of many other genes. The protein, called CREB-binding protein, plays an important role in regulating cell growth and division and is essential for normal fetal development.
1. It is associated with talon’s cusp.
2. Developmental retardation, broad thumb and great toes.
3. Delayed or incomplete descent of testes in males.
110) Sanfilippo’s syndrome :
(Mucopolysaccharidosis III)
MPS-III is a rare autosomal recessive lysosomal storage disease. It is caused by a deficiency in one of the enzymes needed to break down the glycosaminoglycan heparan sulfate.


The four types of MPS-III are due to specific enzyme deficiencies affecting the breakdown of heparan sulfate, which then builds up in various organs. All four types have autosomal recessive inheritance.
Clinical features :
111) Saethre –chotzen syndrome :
(Acrocephalosyndactyly type III )
It is an autosomal dominant trait characterized by short stature and mild mental retardation. congenital disorder associated withcraniosynostosis (premature closure of one or more of the sutures between the bones of the skull). This affects the shape of the head and face, resulting in a cone-shaped head and an asymmetrical face. Individuals with SCS also have droopy eyelids (ptosis), widely spaced eyes (hypertelorism), and minor birth defects of the hands and feet (syndactyly). ] In addition, individuals with more severe cases of SCS may have mild to moderate mental retardation or learning disabilities
Clinical features :
112) Scheie syndrome :
Scheie syndrome is less severe version of Hurler syndrome. It is a condition characterized by corneal clouding, facial dysmorphism, and normal lifespan. People with this condition may have aortic regurgitation
Clinical features :
113) Scalded skin syndrome :
Scalded skin syndrome is caused by infection with certain strains of Staphylococcus bacteria. The bacteria produce a poison that causes the skin damage. The damage creates blisters as if the skin were scalded. Scalded skin syndrome is found most commonly in infants and children under the age of 5.
Clinical features :
114) Scheuthauer-Marie syndrome :
(Cleidocranial dysplasia)
Also known as Marie and Sainton's disease or Scheuthauer—MarieSainton syndrome or Mutational Dysostosis. It is a congenital disorder of autosomal dominant inheritance affecting both sexes equally. Mutations in the core binding factor Alpha-1 gene located on chromosome 6p21 have been suggested to be the cause of CCD. Rearrangement of long arm of chromosome numbers 6 and 8 has also been suggested in favor of CCD. It is also considered to be an autosomal dominant skeletal dysplasia caused by mutations in the bone/cartilage specific osteoblast transcription factor RUNX2 gene.
The most striking features of this syndrome are partial or complete absence of clavicles causing unusual mobility of the shoulders (seen in about 10% of cases) and late closure of fontanelles resulting in frontal bossing. Prolonged retention of deciduous teeth with delay in eruption of succedaneous teeth is the characteristic oral finding. Involvement of facial bones, altered eruption patterns and presence of multiple supernumerary teeth Clinical features:
115) Senear –Usher syndrome:
(Pemphigus erythematosus)
It is an overlap syndrome with features of lupus erythematosus (LE) and pemphigus foliaceus. Pemphigus is demonstrated by acantholysis and immunoglobulin deposits in the interkeratinocyte substance. Patients with pemphigus erythematosus present with vesiculobullae or superficially eroded lesions, which may ooze and crust, particularly in sun-exposed areas, such as the face, the upper part of the chest, and the back. Pemphigus erythematosus may occur at any age, but it is unusual in children. Generally no difference in occurrence of pemphigus erythematosus between the 2 sexes.
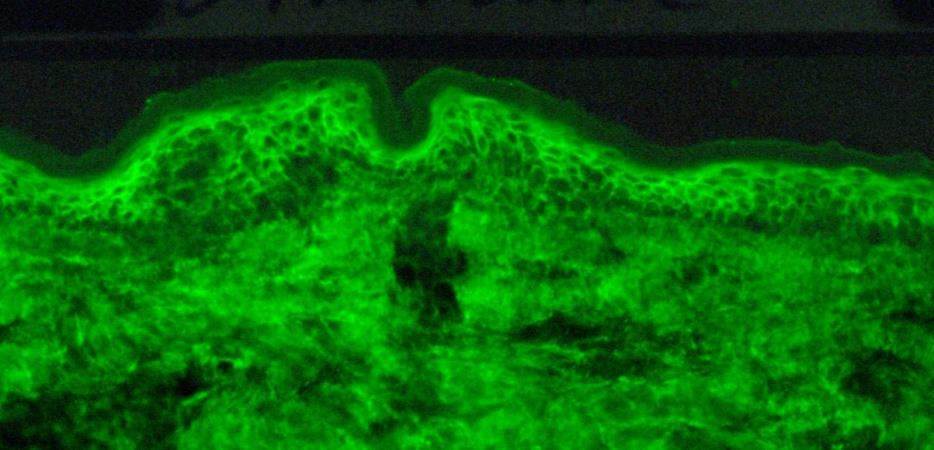
Fig: 39
Direct immunofluorescence microscopy performed on epithelial biopsy specimen obtained from a patient with pemphigus vulgaris detects immunoglobulin G deposits at the epithelial cell surfaces.
116) Silver Syndrome:
(Russell-Silver Syndrome)
Etiology of Russell – silver syndrome is unknown but research points toward a genetic component, possibly following maternal genes. The syndrome is usually caused by a maternal uniparental disomy (UPD) on chromosome 7, in 10% of the cases, which is an imprinting error where the person receives two copies of chromosome 7 from the mother. syndrome' a diagnosis is typically given for children upon confirmation of the presence of several 'symptoms' listed below.
Symptoms are Intrauterine Growth Restriction (IUGR) combined with some of the following:
Oral features :
Fig: 40
117) Sjogren’s Syndrome :
(Sicca syndrome, Gougerot-Mikulicz-Sjogren syndrome, Secreto-inhibitor syndrome)
It was first described in detail by Henrek Sjogren in 1933. Sjogren’s syndrome is a condition originally described as a triad consisting of –
Primary Sjogren’s syndrome (SS) affects the exocrine glands only, primarily the lacrimal and salivary glands. Secondary SS consists of lacrimal and salivary gland involvement with an associated connective tissue disease (like systemic lupus erythematosus, polyarteritis nodosa, scleroderma and rheumatoid arthritis).
Etiology :
Various causes have been suggested such as genetic, hormonal, infectious and immunologic. Now most authors suggest autoimmunity to be the cause, as 75% of the patients had in their sera anti-salivary duct antibody (study by Bertoam). A similar antibody was found in the sera of 24% of patients with systemic lupus erythematosus. The trigger for abnormal immune response is unknown, but some investigators believe that viruses particularly EB or Type A retrovirus, may play an initiating role.
Clinical features :
It occurs more commonly in females after 40 years of age, although children or young adults may be affected. The female:male ratio is around 10:1.
a) Oral manifestations :
Xerostomia or dryness of mouth was the chief complaint of about 82% of patients (in a study by Daniels and colleagues) but the history of salivary gland varies. The dry mouth may be accompanied by bilateral enlargement of parotid glands, unilateral enlargement or no enlargement. Enlargement of sub mandibular glands may also occur. Absence of salivary gland enlargement does not exclude SS as a possible cause of xerostomia. Because of dryness of mouth, patient complains of inability to chew, swallow or wear dentures. There is severe burning sensation of oral mucosa. The lack of oral secretion may lead to secondary oral diseases such as candidiasis or an increase in dental caries. The color of mucosa may vary from pale pink to fiery red. The tongue may be depapillated and so appears smooth and lobulated.
b) Eyes :
c) Mucous membranes :
Dryness of larynx, pharynx and nose is noted by some patients. Lack of secretions in upper respiratory tract may lead to pneumonia. There can also be dryness of vagina.
d) Other symptoms :
The signs of secondary Sjogren syndrome depend chiefly on associated collagen disease and include a wide variety of joint, muscle and skin findings seen in rheumatoid arthritis, scleroderma and systemic lupus erythematosus. Characteristic generalized manifestations of primary SS include renal involvement, polyneuropathy, vasculitis and pneumonitis.
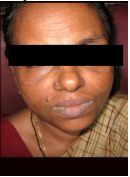

Fig: 41
Photograph depicting dryness of lips (Pics courtesy : Dr. Akhilanand Chaurasiua)
118) Stevens-Johnson Syndrome :
Etiology :
The etiology is unknown. The most common precipitating agent discussed by Shelley is herpes simplex infection, preceding the disease by 1 to 3 weeks. Certain bacterial and fungal infections and certain drugs like barbiturates, phenylbutazone, penicillin etc. may also trigger the disease.
Clinical features :
It is a severe, bullous form of erythema multiforme, with wide spread involvement of skin, oral cavity, eyes and genitalia. It affects males more frequently than females and occurs in young adults.
a) Skin manifestations :
It is characterized by occurrence of asymptomatic, vividly erythematous discrete macules, papules or vesicles and bullae distributed in symmetrical pattern over the hands, arms, feet, leg, face, and neck. The individual lesions may vary in size and are generally a few centimeters less in diameter. A concentric ring-like appearance of the lesions, resulting from the varying shades of erythema, giving rise to ‘target’, ‘iris’, or ‘bull’s eye’ appearance, consisting of a central bulla or pale clearing area surrounded by edema and bands of erythema.
b) Oral manifestations :
Oral lesions commonly appear along with skin lesions in approximately 45% of cases. In some cases oral lesions are predominant or single sign of disease.
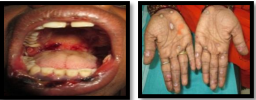
Fig: 42
Oral lesion Target lesions (Bull’s eye)
The oral lesions may be found on lips, buccal mucosa, gingiva, tongue and palate. The initial stage in the development of oral lesion is a small erythematous plaque soon followed by a vesicle or bulla, which ruptures forming shallow erosions covered by necrotic exudates or pseudo membrane. The lips may exhibit ulceration with blo