Chapter Five
5. EMERGING STRUCTURAL INSIGHTS INTO
MULTIDRUG RECOGNITION AND EXTRUSION BY MATE AND MFS TRANSPORTERS
Min Lu*, Katherine Si-Jia Lu
Department of Biochemistry & Molecular Biology, Rosalind Franklin
University of Medicine & Science, North Chicago, Illinois, USA
ABSTRACT
Integral membrane proteins known as multidrug transporters promote multidrug resistance by extruding chemically and structurally dissimilar drugs across the cell membrane, often by utilizing the H+ or Na+ electrochemical gradient. As the current pace of drug discovery is inadequate to address the rapid increase of multidrug resistance, multidrug transporters pose urgent and catastrophic threats to human health by diminishing the efficacy of existing and new therapeutic drugs. Despite such clinical relevance, currently we still lack a deep and mechanistic understanding of how multidrug transporters recognize and extrude different therapeutic drugs. The recent structure determination of several substrate-bound H+- and Na+-dependent multidrug transporters has offered an excellent opportunity to better understand the underlying mechanism of multidrug extrusion. This chapter provides an updated account of the X-ray structures of substrate-bound MATE (Multidrug and Toxic Compound Extrusion) and MFS (Major Facilitator Superfamily) multidrug transporters, as well as their mechanistic implications. Beyond discussing the molecular basis of multidrug recognition and extrusion by MATE and MFS transporters, this chapter may engender new thoughts and ideas about how to curtail efflux-mediated multidrug resistance.
Keywords: multidrug resistance, multidrug extrusion, multidrug transporters, substrate recognition, cation coordination, transport mechanism
*Direct all correspondence to Dr. Min Lu, Department of Biochemistry and Molecular Biology, Rosalind Franklin University of Medicine and Science, 3333 Green Bay Road, North Chicago, Illinois 60064, USA. E-mail: min.lu@rosalindfranklin.edu.
5.1. INTRODUCTION
The ongoing explosion of multidrug resistance continues to plague global and US health care (Higgins, 2007; Fischbach and Walsh, 2009). Multidrug-resistant infectious diseases alone claim more than 2 million lives and cost more than $170 billion per year (Hall et al., 2018). Our arsenal of new therapeutic drugs is being outstripped by the increase in drug resistance (Fischbach and Walsh, 2009).
For instance, no new approved antibiotic class has been discovered over the past 40 years, signaling the worst drought in antibiotic discovery since 1928 (Hall et al., 2018). Needless to say, the public-health crisis caused by multidrug resistance cannot be solved without a deep knowledge of the underlying mechanisms.
Multidrug efflux has emerged as a major mechanism underlying multidrug resistance (Higgins, 2007). Integral membrane proteins known as multidrug transporters curtail the efficacy of existing and new therapeutics by extruding them across the cell membrane (Saier and Paulsen, 2001; Chitsaz and Brown, 2017). Multidrug transporters pose catastrophic and growing threats to humankind because they confer multidrug resistance to pathogenic microorganisms and human cells, making the associated diseases untreatable (Hall et al., 2018).
Thus far, seven families of multidrug transporters have been discovered: namely the ABC (ATP-Binding Cassette), AbgT (p-Aminobenzoyl-glutamate Transporter), DMT (Drug/Metabolite Transporter), MATE (Multidrug and Toxic Compound Extrusion), MFS (Major Facilitator Superfamily), PACE (Proteobacterial Antimicrobial Compound Efflux), and RND (ResistanceNodulation-Division) families (Saier and Paulsen, 2001; Chitsaz and Brown, 2017; Ahmad et al., 2018; Ahmad et al., 2020).
Nearly half of the membrane transport proteins known to date are from the ABC and MFS protein families (Chitsaz and Brown, 2017). The ABC multidrug transporters are “primary” active membrane transport proteins powered by ATP hydrolysis (Saier and Paulsen, 2001). By contrast, the DMT, MATE, MFS and RND families of multidrug transporters are “secondary” active transporters that utilize the transmembrane H+ or Na+ electrochemical gradient to translocate drugs (Chitsaz and Brown, 2017).
The DMT, MFS, and RND multidrug transporters typically employ the H+ gradient to extrude drugs, whereas the MATE transporters use either the H+ or Na+ gradient (Chitsaz and Brown, 2017). Based on amino-acid sequence similarity, the ~900 MATE transporters can be classified into the NorM, DinF (DNA damage-inducible protein F) and eukaryotic subfamilies (Brown et al., 1999; Omote et al., 2006; Kuroda and Tsuchiya, 2009; Lu, 2016).
Members of the NorM and DinF subfamilies can utilize the H+ or Na+ electrochemical gradient, whereas the eukaryotic MATE proteins are H+-dependent (Morita et al., 2000; He et al., 2004; Otsuka et al., 2005a; for additional references see Lu, 2016). Both the NorM and DinF subfamilies contain eubacterial and archaeal members, which share rather modest amino-acid sequence similarity (Lu, 2016). MATE transporters can extrude polyaromatic drugs that carry positive charges at physiological pH, although these drugs exhibit rather diverse chemical structures and properties (Omote et al., 2006; Kuroda and Tsuchiya, 2009).
The more than 1 million sequenced MFS proteins, on the other hand, comprise the largest family of solute transport proteins and at least 82 subfamilies (for review see Saier and Paulsen, 2001; Yan, 2013; Quistgaard et al., 2016). Previous studies of MFS transporters such as LacY (lactose permease) and GLUTs (glucose transporters), which are from the Oligosaccharide/H+ Symporter (OHS) and Sugar Porter (SP) subfamilies, respectively, revealed the shared MFS transporter architecture and provided critical insights into the transport mechanism (for review see Kaback and Guan, 2019; Yan, 2017).
MdfA, a polyspecific MFS multidrug transporter from the ubiquitous Drug/H+ Antiporter-1 (DHA1) subfamily, can export physicochemically disparate compounds that lack a common chemical structure (Edgar and Bibi, 1997; for review see Fluman and Bibi, 2009; Yardeni et al., 2017). Previous studies suggested that MdfA is mechanistically distinct from the substrate-specific MFS transporters such as LacY, likely representing a new paradigm for membrane transport proteins (Fluman et al., 2012; Tirosh et al., 2012; Fluman et al., 2014).
Unfortunately, we are now on the losing side of the war against multidrug resistance because the current pace of drug discovery is woefully inadequate to tackle multidrug efflux (Fischbach and Walsh, 2009). This doomsday scenario cannot be averted unless we understand how multidrug transporters extrude drugs. At the present time, we do not fully understand how these drug-exporting proteins select their substrates, how they move therapeutic drugs across the cell membrane, or how they can be countervailed for potential therapeutic benefit (Lu, 2016).
Over the past 6 years, major strides have been made toward illuminating how MATE and MFS multidrug transporters recognize and extrude drugs, but these new mechanistic insights have yet to be reviewed collectively (Lu, 2016; Wu et al. 2019). This chapter summarizes these recent findings, focuses on what has been learned from the crystal structures of substrate-bound MATE and MFS multidrug transporters (Lu et al., 2013a; Lu et al., 2013b; Wu et al., 2019), and asks new questions that need to be addressed by future work.
5.2. THE SUBSTRATE-BOUND STRUCTURE OF NORM-NG
The X-ray structures of at least eight MATE transporters have been published, including NorM from Vibrio cholerae (NorM-VC; He et al., 2010), NorM from Neisseria gonorrhoeae (NorM-NG; Lu et al., 2013b), DinF from Pyrococcus furiosus (PfMATE; Tanaka et al., 2013), DinF from Bacillus halodurans (DinF-BH; Lu et al., 2013a), DinF from Escherichia coli (ClbM; Mousa et al., 2016), DinF from Vibrio cholerae (VcmN; Kusakizako et al. 2019), eukaryotic MATE transporter from Camelina sativa (CasMATE; Tanaka et al. 2017), and eukaryotic MATE protein from Arabidopsis thaliana (AtDTX14; Miyauchi et al., 2017).
Among them, the drug-bound structures of only three MATE transporters, NorM-NG, PfMATE and DinF-BH, have been reported. Since the function of PfMATE is questionable (Zakrzewska et al., 2019), the mechanistic implications of the drug-bound PfMATE structure remain controversial (Lu, 2016) and hence is excluded from this chapter.
The first substrate-bound structure of any MATE transporter, i.e., that of NorM-NG, captures the transporter in an outward-facing, substrate-bound state (Lu et al., 2013b). NorM-NG traverses the membrane bilayer twelve times giving rise to twelve membrane-spanning segments (TM1-TM12). Near the middle of the lipid bilayer, similarly folded N (TM1-TM6) and C (TM7-TM12) domains point away from one another toward the periplasm, yielding a V-shaped conformation (Figure 1a).
TM1-TM12 are connected by eleven intracellular and extracellular loops, denoted L1-2 to L11-12. Among them, L3-4, L9-10 and L6-7 are conspicuously long (Lu et al., 2013b). The intracellular L6-7 delimits the N and C domains of NorM-NG, whereas the extracellular L3-4 and L9-10 insert into a cavity formed between the N and C domains.
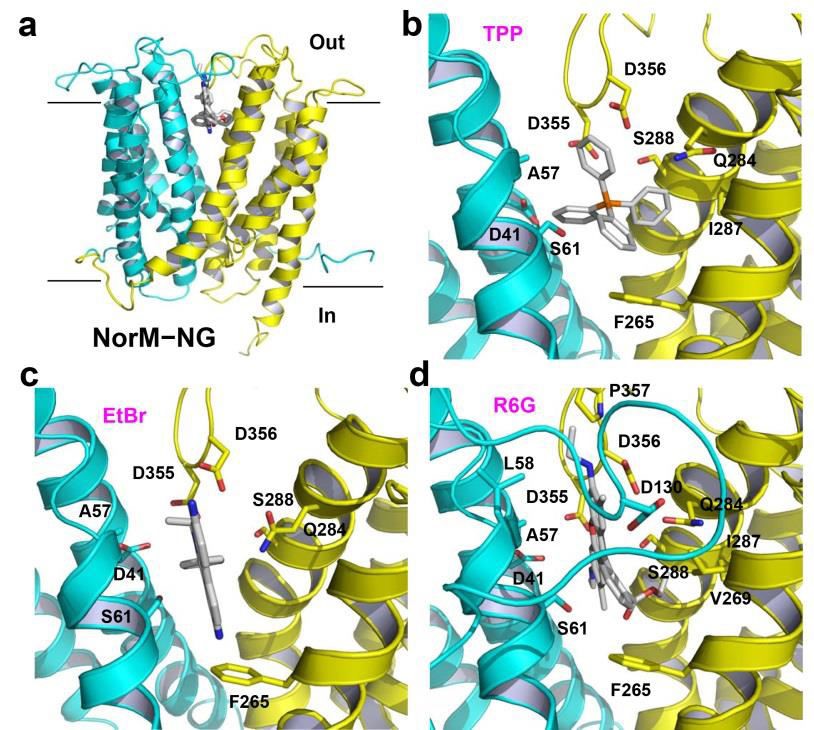
Figure 1. Structure of the multidrug-binding site in NorM-NG. (a) NorM-NG is drawn as a ribbon and viewed from the membrane plane, whereas the three substrates are in stick representation and colored grey. The N (residues 5-230) and C (residues 231-459) domains of NorM-NG are colored cyan and yellow, respectively. (b),(c),(d) Close-up of the binding site for TPP (PDB 4HUK), ethidium (PDB 4HUM), and R6G (PDB 4HUN), respectively. Relevant amino acids are illustrated as sticks. IL3-4 was omitted in (b) and (c) for clarity.
The bottom of this cavity is situated about half way through the membrane and is defined by D41, S61, F265 and I292, which are from TM1, TM2, TM7 and TM8, respectively (Lu et al., 2013b). The cavity is sealed from the cytoplasmic side by highly ordered protein structure, ~20 Å thick. The wall of the cavity is formed by T42, A57, L58, V269, Q284, V285, I287 and S288; and the extracellular loops L3-4 and L9-10, which donate S129, D130, D355, D356 and P357 into the cavity.
Except for D130, V269 and P357, all the amino acids surrounding this cavity are conserved or semi-conserved in the NorM subfamily. The interior of the cavity exhibits a surplus of negative charges, suggesting potential electrostatic attraction for cations or zwitterions (Lu et al., 2013b).
5.3. THE MULTIDRUG-BINDING SITE IN NORM-NG
Since NorM-NG was crystallized in the absence of added monovalent cations including Na+, the TPP-, ethidium- and R6G-bound NorM-NG structures were interpreted to depict a Na+-free, substrate-bound state of the transporter (Lu et al., 2013b). Each of the three substrates buries ~70% of accessible surface area upon binding NorM-NG, which is similar to values obtained for multidrug-binding transcription factors (Bachas et al., 2011; Newberry et al., 2008). Also akin to these water-soluble transcription factors, the docking locations for TPP, ethidium and R6G are similar in NorM-NG, which are near the membrane-periplasm interface (Lu et al., 2013b).
Numerous interactions were observed between NorM-NG and the bound substrates (Lu et al., 2013b). In particular, the side chains of three acidic amino acids, D41, D355 and D356, make long-range electrostatic interactions with the bound substrates (Figures 1b,c,d). The distances between the side-chain carboxyl groups and the positively charged atoms in the substrates are from 3.4 to 7.2 Å (Lu et al., 2013b).
Moreover, the side chains of S61, Q284 and S288 also contribute to charge complementation, which make charge-dipole interactions with the bound substrates, because the side-chain hydroxyl and carbonyl groups are within 5 Å of the positively charged atoms in the substrates. Surprisingly, the hydrophobic interactions between NorM-NG and the bound substrates are scarce, involving only A57 and F265. F265 in particular makes an edge-to-face aromatic stacking interaction with the substrates (Figures 1b,c,d).
To validate the biological relevance of the multidrug-binding site, the amino acids involved in the binding of all three substrates were substituted. These single mutations, with the exception of S61A, severely impaired the ability of NorM-NG to confer resistance to E. coli against TPP, ethidium or R6G (Lu et al., 2013b). Congruent with these observations, these inactivating mutations exerted deleterious effects on the Na+-dependent drug efflux function.
Altogether, the data suggested that D41, F265, Q284, D355 and D356 play critical roles in multidrug transport, thereby supporting the functional importance of the observed multidrug-binding site. Furthermore, mutations of the counterparts of S266 and S288 in the human MATE protein hMATE1 affected the drug-binding affinity (Otsuka et al., 2005b; Matsumoto et al., 2008), implying a common substrate-binding site among the NorM and eukaryotic MATE transporters (Lu et al., 2013b; Lu, 2016).
Interestingly, in contrast to the multidrug transporters from the ABC, DMT, and MFS families, which utilize numerous aromatic amino acids to bind their substrates (Chen et al., 2007; Aller et al., 2009; Heng et al., 2015), NorM-NG employs a small number of hydrophobic residues to interact with drugs (Lu et al., 2013b).
At least three acidic residues are used by NorM-NG to neutralize the substrates, which may be important for preventing negatively charged or electroneutral compounds from binding and transport by the protein, thus conferring substrate specificity. The paucity of aromatic and non-polar residues may enable NorM-NG to avoid overly tight association with hydrophobic drugs when the transporter is poised to release substrate (Lu et al., 2013b).
In addition, there are no substantial changes in the positions of amino-acid side chains upon binding different substrates in NorM-NG, similar to what had been observed in the multidrug-binding transcription factors (Bachas et al., 2011; Newberry et al., 2008). The multiple acidic residues in NorM-NG may allow versatile orientation and charge complementation of structurally dissimilar cationic drugs without the need to drastically alter the structure of the multidrugbinding site (Lu et al., 2013b).
Additionally, extracellular loops L3-4 and L9-10 cover the multidrug-binding cavity, which may provide flexibility to optimize transporter-substrate interactions. Indeed, it was subsequently discovered that these extracellular loops can undergo structural changes when NorM-NG interacts with different ligands (Radchenko et al., 2015).
5.4. THE CATION-BINDING SITE IN NORM-NG
Besides the Na+-free, substrate-bound structures of NorM-NG, the Cs+-bound structure of NorM-VC was also reported (Lu et al., 2013b). The Cs+-bound structure represents a “drug”-bound, cation-bound NorM-NG wherein an unidentified ligand acts as a substrate surrogate. This co-structure exhibited no significant conformational difference from that of Na+-free, drug-bound NorMNG (rms deviation ~0.5 Å).
This finding is unexpected because the canonical antiport mechanism predicts that the counter-transported cation and substrate compete for a shared binding-site in the transporter, and that substrate and cation must not bind the protein simultaneously (for review see Schuldiner, 2014).
In the Cs+-bound structure of NorM-NG, the carboxyl group of a conserved E261 and the aromatic ring of a conserved Y294 are within 3.2 Å and 4.4 Å of the cation, respectively, implicating E261 and Y294 as the Na+-coordinating residues (Figure 2a). Notably, no substrate-binding amino acids in NorM-NG directly coordinate Cs+, which mimics the counter-transported Na+. Interestingly, Y294 makes an uncommon cation-π interaction (Dougherty, 1996).
Of note, membrane protein-ligand interactions mediated by cation-π interactions usually involve positively charged amines, which had been found in a number of ligand-gated acetylcholine receptors and G-protein-coupled receptors, as well as the betaine and carnitine transporters (for review see Zacharis and Dougherty, 2002; Ressel et al., 2009; Tang et al., 2010; Schulze et al., 2010).
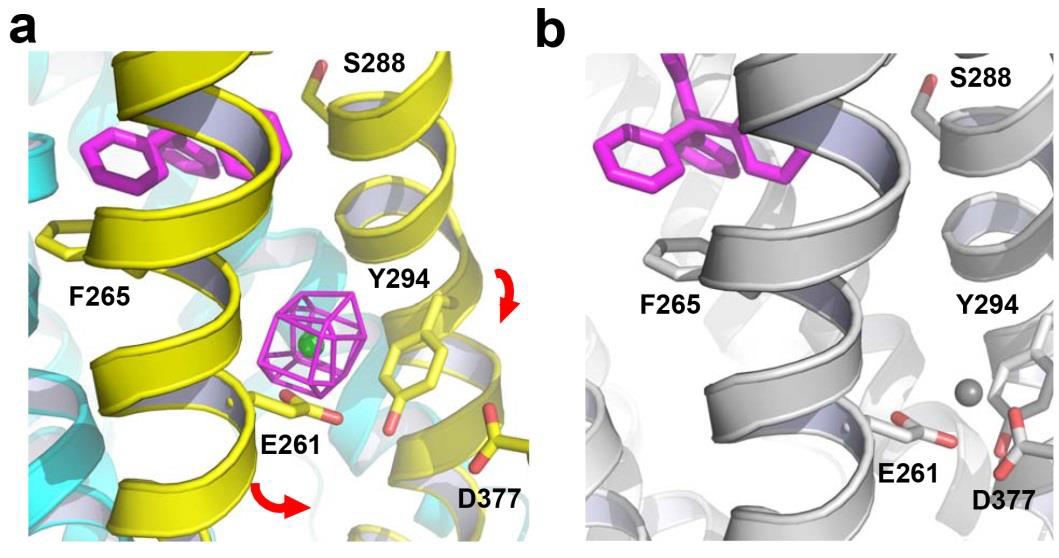
Figure 2. Structure of the cation-binding site in NorM-NG. (a) Cs+-bound structure of NorM-NG (PDB 4HUL). Cs+ (green sphere) is overlaid with difference isomorphous Fourier map (magenta mesh) contoured at 6 σ. Red arrows indicate a proposed movement of TM7 and TM8 towards TM10. TPP (magenta) taken from the TPP-bound structure (PDB 4HUK) is shown in stick representation to indicate the multidrug-binding site. (b) Hypothetical Na+ (gray sphere) coordination arrangement, relevant amino acids are depicted as stick models and NorM-NG is colored gray.
Generally speaking, cation-π interactions are largely electrostatic in nature and thus well-suited for molecular interaction within a hydrophobic environment, including the lipid bilayer (Dougherty, 1996). Nonetheless, a Na+-π interaction had never been observed in any membrane protein prior to structure determination of NorM-NG (Lu et al., 2013b). Y294 in NorM-NG presumably makes a cation-π interaction with Na+, which is also the preferred counter-transported cation (Long et al., 2008).
This cation selectivity likely results from the side-chain carboxylate of E261 being a high-field-strength ligand that favors Na+ over K+, as well as from a higher binding affinity for the Na+-π relative to K+-π interaction (Noskov and Roux, 2008; Remko and Soralova, 2012). Furthermore, the relatively weak Na+-π interaction may offer kinetic advantages, for example, faster rates of Na+-binding and/or unbinding, as compared with the more common Na+-coordination arrangement that exclusively involves oxygen atoms (Harding, 2002; Xue et al., 2008).
Mutation of either E261 or Y294 was found to impair the transport function of NorM-NG, therefore supporting the functional importance of the observed cation-binding site (Lu et al., 2013b). Furthermore, although the distance between the carboxyl group of D377 and Cs+ exceeds 7.2 Å in NorM-NG, previous studies suggested the counterpart of D377 in NorM-VC as a Na+-coordinating residue in a cation-bound, substrate-free state (He et al., 2010; Lu et al., 2013b).
Mutational studies of D377 counterparts in yet another two MATE proteins, NorM-VP and hMATE1, also indicated that D377 plays an important role in multidrug transport (Otsuka et al., 2005; Matsumoto et al., 2008). Thus, it was concluded that D377 likely coordinates Na+ in the substrate-free NorM-NG (Figure 2b).
5.5. THE TRANSPORT MECHANISM OF NORM-NG
Based on available biochemical data and structure comparison between the cation-bound, drug-free NorM-VC and drug-bound NorM-NG, a transport mechanism was proposed (Lu et al., 2013b; Lu, 2016). This mechanism predicts that the outward-facing, drug-bound NorM-NG utilizes E261 and Y294 to initiate Na+-binding from the periplasmic side (Figure 3). Subsequently, D377 becomes involved in Na+-coordination as TM7 and TM8 shift toward TM10.
As a consequence, F265, Q284 and S288 move away from the multidrugbinding site, thereby triggering the release of bound drug. The Na+-bound, drugfree NorM-NG then transitions into the inward-open conformation to capture a new drug. Afterwards, TM7 and TM8 move back towards the central cavity upon drug binding, which drives the release of Na+ into the cytoplasm. The drug-bound, Na+-free NorM-NG finally switches to the outward-facing conformation to start a new transport cycle (Figure 3).
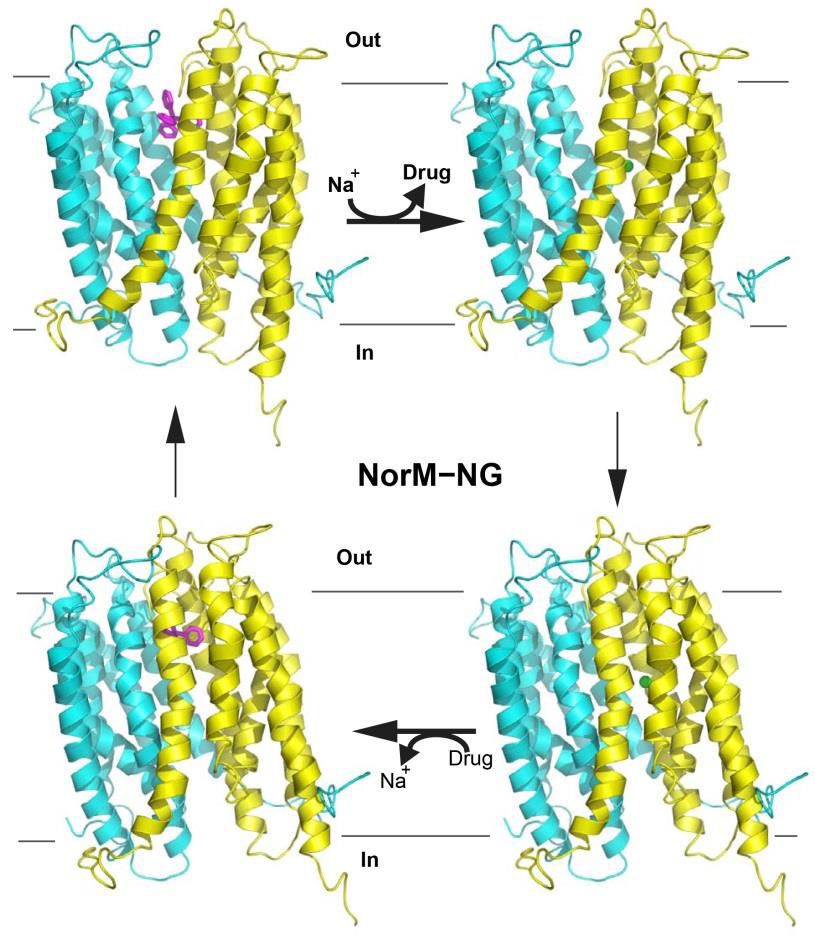
Figure 3. Proposed transport mechanism for NorM-NG. The protein is shown in ribbon representation; TPP (magenta) is drawn as a stick model and Na+ as a green sphere. The N (residues 5-230) and C (231-459) domains of NorM-NG are colored cyan and yellow, respectively. Na+ binding to the outward-facing, drugbound NorM-NG (top left) triggers the release of drug. Subsequently, the Na+bound, outward-facing NorM-NG (top right) then switches to the Na+-bound, inward-facing state (bottom right). Drug binding to NorM-NG promotes the dissociation of Na+ from the transporter to yield the drug-bound, inward-facing state (bottom left), which eventually returns to the drug-bound, outward-facing conformation (top left).
This mechanism suggests that Na+ and substrate alternately bind to two spatially distinct sites in NorM-NG during the transport cycle, rather than competing for a common subset of amino acids (Lu et al., 2013b; Lu, 2016). This non-canonical, indirect competition based antiport mechanism involves a fullyloaded intermediate state in which the substrate and counter-transported cation bind to the transporter simultaneously, as supported by the Cs+-bound NorM-NG structure (Lu et al., 2013b). In this mechanism, the coupling between drug and counter-transported Na+ is mediated by structural changes within the transporter (Lu, 2016).
5.6. THE STRUCTURE OF SUBSTRATE-BOUND DINF-BH
Besides NorM-NG, the substrate-bound structure of DinF-BH was also published (Lu et al., 2013a). In contrast to NorM-NG, which is a Na+-coupled antiporter, DinF-BH is H+-dependent. Moreover, in NorM-NG, the N domain (TM1-TM6) is structurally related to the C domain (TM7-TM12) around a pseudo-twofold rotational symmetry axis normal to the membrane plane, and the structures of the two domains can be superimposed onto each other with an rms deviation of 2.4 Å (Lu et al., 2013b).
By contrast, structural superimposition of the two domains in DinF-BH results in an rms deviation of 3.3 Å, with the largest difference being in the extracellular halves of TM7-TM8 and their counterparts in TM1-TM2 (Lu et al., 2013a). If those protein residues are excluded, the rms deviation can be lowered to 2.5 Å. Of note, TM7 and TM8 in DinF-BH are more bent near their midsections than TM1 and TM2, respectively.
The bending of TM7 and TM8 in DinF-BH creates a crevice in the C domain (Figure 4a), which is accessible to the periplasm and outer membrane leaflet (Lu, et al. 2013a). This crevice separates the C domain into two layers, with TM7 and TM8 in one and TM9 to TM12 in the other. This helical arrangement suggests that in order to form a similar cation-binding site to that of NorM-NG (Lu et al., 2013b), the unbending or straightening of both TM7 and TM8 is required (Lu et al., 2013a).
In addition, the DinF subfamily lacks the cation-binding motif identified in NorM-NG, which involves E261 (TM7), Y294 (TM8) and D377 (TM10). These data implied that the cation-binding site of DinF-BH differs from that of NorMNG (Lu, 2016). Indeed, further structural and biochemical studies revealed that the drug/cation coupling mechanism is different between NorM-NG and DinF-BH (Lu et al., 2013a; Radchenko et al., 2015).
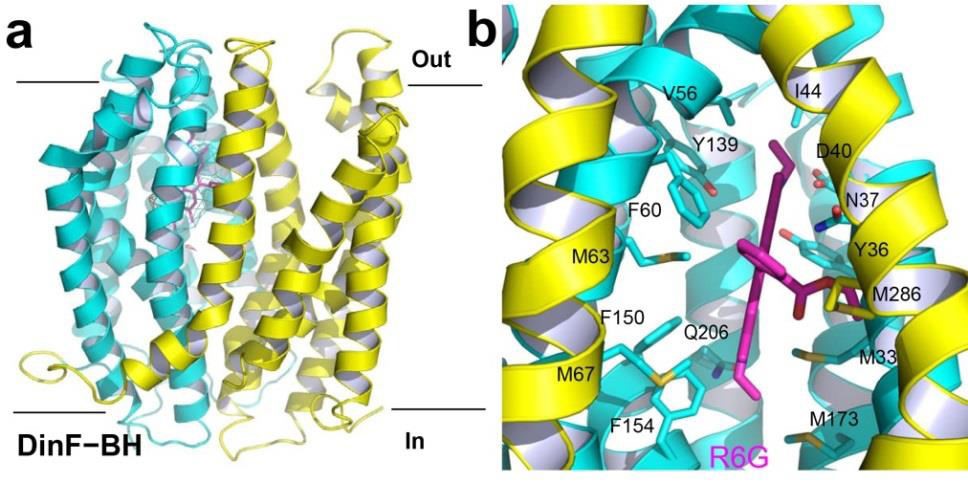
Figure 4. Structure of the substrate-binding site in DinF-BH. (a) DinF-BH is shown in ribbon rendition as viewed from the membrane plane, with residues 3227 and 228-448 colored cyan and yellow, respectively (PDB 4LZ9). The bound R6G is drawn as magenta sticks, which is overlaid with the experimental electron density map (cyan mesh) contoured at 1.5 σ. (b) Close-up of the drug-binding chamber, R6G (magenta) and relevant amino acids are displayed in stick representation.
In DinF-BH, TM1-TM8 collectively forms an internal chamber that shelters the bound substrate R6G (Lu et al. 2013a). This chamber harbors numerous hydrophobic amino acids, whose bulky side chains can shield R6G from the external environment (Figure 4b). Additionally, TM7 and TM8 define a lateral opening through which protons from the solvent-filled crevice can enter the chamber.
At the widest point within the membrane bilayer, the opening is 10 Å wide, which is spatially sufficient to allow R6G to diffuse out of the chamber and into the crevice (Lu et al., 2013a). The access to the chamber from the cytoplasmic side, however, is blocked by highly ordered protein structure, suggesting that the R6G-bound structure captures DinF-BH in an outward-facing state.