Chapter Eight
8. CLONING, AMPLIFIED EXPRESSION,
FUNCTIONAL CHARACTERISATION AND PURIFICATION OF VIBRIO PARAHAEMOLYTICUS
NCS1 CYTOSINE TRANSPORTER VPA1242
Irshad Ahmad1, Pikyee Ma2, Nighat Nawaz3, David J.
Sharples, Peter J. F. Henderson*, Simon G. Patching*
School of BioMedical Sciences and Astbury Centre for Structural Molecular
Biology, University of Leeds, Leeds, LS2 9JT, UK
ABSTRACT
The Nucleobase Cation Symporter-1 (NCS1) family of secondary active transport proteins comprises over 2,500 sequenced members from bacteria, archaea, fungi and plants. In bacteria NCS1 transporters function in salvage pathways for nucleobases and nucleosides, hydantoins and other related compounds. To-date only three bacterial NCS1 proteins are experimentally characterised: Mhp1 (5-arylhydantoins), PucI (allantoin), CodB (cytosine). In this work we cloned the gene for Vibrio parahaemolyticus protein VPA1242 (412 residues) into plasmid pTTQ18 with concomitant introduction of a C-terminal hexahistidine-tag and achieved amplified expression in Escherichia coli BL21(DE3). VPA1242 is predicted to contain twelve transmembrane spanning 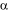 -helices with both N- and C-terminal ends in the cytoplasm and it shares high sequence homology with CodB (75.2% identical plus 14.3% highly similar residues). In transport assays, VPA1242 mediated uptake of 3H-cytosine into energised whole cells and no significant uptake of any other tested radiolabelled compounds. 3H-cytosine uptake was not dependent on sodium, suggesting that VPA1242 transport is driven by a proton gradient. Based on competition of 3H-cytosine uptake by unlabelled compounds, VPA1242 was highly specific for cytosine. The only other compounds that had small but significant competitive effects were hydantoin, benzyhydantoin, and uracil, which share some structural similarity to cytosine. VPA1242 was stable to detergent solubilisation and purification using a Ni-NTA resin, achieving a purity of ~85% and a yield of ~0.9 mg/litre from fermentor cultures, and had stable -helical content, all making it tractable to further structural and biophysical characterisation.
-helices with both N- and C-terminal ends in the cytoplasm and it shares high sequence homology with CodB (75.2% identical plus 14.3% highly similar residues). In transport assays, VPA1242 mediated uptake of 3H-cytosine into energised whole cells and no significant uptake of any other tested radiolabelled compounds. 3H-cytosine uptake was not dependent on sodium, suggesting that VPA1242 transport is driven by a proton gradient. Based on competition of 3H-cytosine uptake by unlabelled compounds, VPA1242 was highly specific for cytosine. The only other compounds that had small but significant competitive effects were hydantoin, benzyhydantoin, and uracil, which share some structural similarity to cytosine. VPA1242 was stable to detergent solubilisation and purification using a Ni-NTA resin, achieving a purity of ~85% and a yield of ~0.9 mg/litre from fermentor cultures, and had stable -helical content, all making it tractable to further structural and biophysical characterisation.
* Direct all correspondence to Prof. Simon Patching or Prof. Peter Henderson, Astbury Building, Faculty of Biological Sciences, University of Leeds, Leeds, LS2 9JT, UK. E-mail: s.g.patching@leeds.ac.uk, simonpatching@yahoo.co.uk, p.j.f.henderson@leeds.ac.uk. Current addresses:
Keywords: bacteria, cloning strategy, CodB, cytosine, NCS1, nucleobase cation symporter, protein purification, substrate specificity, transport protein
8.1. INTRODUCTION
The Nucleobase Cation Symporter-1 (NCS1) family of secondary active transport proteins comprises over 2,500 sequenced members derived from Gramnegative and Gram-positive bacteria, archaea, fungi and plants (de Koning and Diallinas, 2000; Pantazopoulou and Diallinas, 2007; Saier et al., 2009; Weyand et al., 2010; Witz et al., 2014; Krypotou et al., 2015; Ma et al., 2016; Sioupouli et al., 2017; Patching, 2018). These proteins generally function in salvage pathways as transporters for purine and pyrimidine nucleobases and nucleosides, hydantoins and other related compounds including pyridoxine, thiamine and uric acid. NCS1 family proteins are usually 419-635 amino acid residues in length and are usually predicted to possess twelve transmembrane spanning α-helices (Saier et al., 2009; Witz et al., 2014) and function using a symport mechanism driven by a proton or sodium gradient (Krypotou et al., 2015). The common transport mechanism catalysed by NCS1 family proteins is simplified as: Nucleobase or Hydantoin or Vitamin (out) + H+ or Na+ (out) → Nucleobase or Hydantoin or Vitamin (in) + H+ or Na+ (in). NCS1 proteins show no sequence similarity to the NCS2 family, also known as the Nucleobase Ascorbate Transporter (NAT) family (Goudela et al., 2005; Gournas et al., 2008; Diallinas and Gournas, 2008; Frillingos, 2012). The first structural model for the NCS1 family was the sodium-coupled hydantoin transport protein, Mhp1, from Microbacterium liquefaciens (Suzuki and Henderson, 2006; Jackson et al., 2013; Patching, 2017; Patching, 2018), for which crystal structures have been determined with the protein in three different conformations i.e. outward-facing open, occluded with substrate and inwardfacing open (Weyand et al., 2008; Shimamura et al., 2010; Simmons et al., 2014). Mhp1 has provided a principal model for the alternating access mechanism of membrane transport and for the mechanism of ion-coupling (Shimamura et al., 2010; Weyand et al., 2011; Adelman et al., 2011; Shi, 2013; Kazmier et al., 2014; Kazmier et al., 2017). The function and substrate specificities of only 25 NCS1 proteins (3 bacterial, 16 fungal, 6 plant) have so far been characterised experimentally (Schwacke et al., 2003; Krypotou et al. 2015; Patching, 2018). The two other characterised bacterial NCS1 proteins are allantoin transporter PucI from Bacillus subtilis (Ma et al., 2016) and cytosine transporter CodB from Escherichia coli (Danielsen et al., 1995).
Studies suggest that the two distinct fungal NCS1 subfamilies and the plant homologues originated through independent horizontal transfers from prokaryotes and demonstrate that transport activities in NCS1 proteins appeared independently by convergent evolution (Hamari et al., 2009; Krypotou et al., 2015). This is one explanation for the observation that substrate specificities of (fungal and plant) NCS1 proteins cannot be predicted by simple amino acid sequence comparisons, by phylogenetic analyses or from comparisons of amino acid residues in the major substrate binding site. Indeed, comparisons performed with Fur-type and Fcytype NCS1 fungal transporters showed how identical or highly similar residues can provide different substrate specificities due to convergent evolution within the major substrate binding site (Hamari et al., 2009; Krypotou et al., 2015). Further investigations into the origins of substrate specificity in the wider NCS1 family are therefore warranted, especially those from bacteria. In this work we present results for cloning, amplified expression, functional characterisation and purification of NCS1 transport protein VPA1242 from Vibrio parahaemolyticus (UniProt entry Q87GS3).
V. parahaemolyticus is a Gram-negative marine bacterium and is a worldwide leading cause of foodborne gastroenteritis in humans, especially in the areas with high consumption of raw or undercooked seafoods (Letchumanan et al., 2014; Letchumanan et al., 2015). The organism is naturally found in estuarine, marine and coastal environments and possesses enormous adaptive capabilities: a planktonic cell or attached to submerged inert surfaces, such as the underside of boats or to other ocean surfaces like fish, shellfish and zooplankton (McCarter, 1999; Iwamoto et al., 2010; Nelapati et al., 2012). V. parahaemolyticus exists either as a swimmer cell or a swarmer cell, adapted for locomotion in various environments (Sar et al., 1990; Makino et al., 2003). Under appropriate circumstances the organism has an exceptionally short generation time of 8-12 minutes (Ulitzur, 1974; Makino et al., 2003). A haemolysin is thought to be an important virulence factor produced by the bacterium but the mechanisms of pathogenesis are still unclear (Hiyoshi et al., 2010; West et al., 2013; Lee et al., 2015; Kumaran and Citarasu, 2016). The only predicted NCS1 family transporter in V. parahaemolyticus is VPA1242, which is closely related to cytosine transporter CodB from E. coli (Danielsen et al., 1995).
For investigating V. parahaemolyticus protein VPA1242, we used a wellestablished strategy for cloning bacterial transport proteins with a C-terminal hexahistidine tag in E. coli using plasmid pTTQ18, followed by optimisation of conditions for amplified expression. The functional activity of VPA1242-His6 was characterized in terms of substrate and ion specificity and ligand recognition. VPA1242-His6 was purified by immobilised metal affinity chromatography (IMAC) with a Ni-NTA resin, followed by analysis of secondary structure integrity and thermal stability using circular dichroism spectroscopy.
8.2. MATERIALS AND METHODS
8.2.1. General
Chemicals, reagents and media of the highest available quality were obtained from Sigma-Aldrich Co., Fisher Scientific UK Ltd, Melford Laboratories Ltd, BDH Chemical Supplies or Difco Laboratories, unless stated otherwise. All media, buffers and other solutions were prepared using either deionised water or MilliQTM water. All media were sterilised by autoclaving or for thermallysensitive solutions by passage through 0.2 μM Minisart® high-flow sterile syringe-driven filters (Sartorius) or using vacuum-driven 0.2 μM filters (Stericup®) from Millipore. Cellulose nitrate 25 mm ø filters (0.45 μM pore size) for radiolabelled substrate assays and cellulose ester GSTF 25 mm ø filters (0.22 μm pore size) (Whatman®) for protein determinations were from Millipore (UK)
Ltd. DNA purification kits were from QIAGEN Ltd. Restriction endonucleases and T4 DNA ligase were from New England Biolabs, Pfu TurboTM DNA polymerase was from Agilent Technologies UK, and 1 kb DNA ladder and SYBR SafeTM DNA gel stain was from Invitrogen. PCR amplification of DNA was performed using a Peltier Thermal cycler from MJ Research. Cell disruption was performed using a Constant Systems disruptor. Protein determinations used the method of Schaffner and Weissmann (1973) or a BCA assay using Pierce® BCA protein assay reagent A from Thermo Scientific. SDS-PAGE was performed by the method of Laemmli (1970), refined for membrane proteins as described by Henderson and Macpherson (1986) using 4% stacking gels and 15% resolving gels in a BioRad Mini PROTEAN 3 apparatus. Acrylamide (40%) and bisacrylamide (2%) solutions were from BioRad Laboratories and SDS-7 protein molecular weight markers were from Sigma-Aldrich Co. Western blotting was performed by semi-dry transfer using a BioRad TRANS-BLOT® SD apparatus; RGS-His6 antibody was from QIAGEN Ltd, SuperSignal® West Pico luminal enhancer solution and stable peroxide solution were from Perbio Science UK and FluorotransTM membrane was from Pall BioSupport, UK. High-range Rainbow molecular weight markers were from Amersham Biosciences UK Ltd.
8.2.2. Gene cloning and amplified expression
Cloning and amplification of expression of V. parahaemolyticus protein VPA1242 in E. coli was achieved using a strategy that we and others have found successful with a wide range of bacterial and archaeal membrane proteins (Ward et al., 1999; Saidijam et al., 2003; Saidijam et al., 2005; Clough et al., 2006; Suzuki and Henderson, 2006; Szakonyi et al., 2007; Gordon et al., 2008; Ma et al., 2008; Bettaney et al., 2013; Ma et al., 2013; Ma et al., 2016). This involved design of PCR primers (Forward: 5ꞌ-CCGGAATTCGCATATGGCTGGAGACAATAACTACAGTCTTGGACCAG-3ꞌ; Reverse: 5ꞌ-AAAACTGCAGCC GCT
GGCTGTGAAGCCAGTGCTTTTTTGTTGAG-3ꞌ) to extract and amplify the specific gene from genomic DNA of V. parahaemolyticus, introducing EcoRI and PstI restriction sites at the 5ꞌ and 3ꞌ ends, respectively. The restriction-digested PCR product was ligated into plasmid pTTQ18 (Stark, 1987) immediately upstream from a hexahistidine (His6) tag coding sequence and the resultant pTTQ18-gene(His6) construct was used to transform E. coli BL21(DE3) cells. Expression tests were initially performed using small scale cultures (50 ml) in LB medium supplemented with carbencillin (100 µg/ml), induction with IPTG (0.5 mM) at A600 = 0.6 and then growth for a further 2 hours. Total (inner plus outer) membranes from induced and uninduced cells were isolated using the water lysis procedure (Witholt et al., 1976; Ward et al., 2000) and analysed by SDS-PAGE and Western blotting using an antibody to the His6 epitope for detection of amplified protein bands. Conditions for optimising the amplified expression level of VPA1242-His6 were tested by using ranges in the concentration of IPTG for induction (0-1 mM) and the length of induction (2-5 hours) and using three different types of media for cell growth (LB, 2TY, M9 minimal).
8.2.3. Scale up and membrane preparation
To provide sufficient quantities of protein for purification and further analysis, optimised culture conditions were scaled up to volumes of 10 litres in flasks or 30 litres in a fermentor. Typically a total of 10 litres of cells were grown to an A600 of 0.4-0.6, then induced with IPTG (0.5 mM) and grown for a further 3 hours before harvesting by centrifugation (6000 x g, 15 min, 4 °C) and storage at 80 °C. At a later time the cells were thawed, suspended in Tris-EDTA buffer (20 mM Tris, pH 7.5 with 0.5 mM EDTA) and disrupted by passing twice through a cell disrupter (Constant Systems) at 30 kpsi. Undisrupted cells and cell debris were removed by centrifugation at 12000 x g for 45 minutes at 4 °C. The supernatant containing total (inner plus outer) membranes was collected and retained. Inner/outer membranes were separated by sucrose gradient ultracentrifugation and prepared as described in Ward et al. (2000), followed by washing and resuspension in Tris buffer (20 mM, pH 7.5), dispensing into aliquots, rapid freezing in liquid nitrogen and storage at -80 °C.
8.2.4. Whole cell transport and competition assays
Measurements of uptake of radiolabelled compounds into energised whole cells of E. coli were performed using a method based on that of Henderson et al. (1977). Radiolabelled compounds were synthesised in-house (Patching, 2009; Patching, 2011; Patching, 2017) or from Perkin Elmer Ltd, UK. Cells were grown in LB medium supplemented with glycerol (20 mM) and carbenicillin (100 μg/ml) in volumes of 50 or 100 ml at 37 °C in 250 ml or 500 ml baffled conical flasks with aeration at 220 rpm to an A600 of ~0.8. The cells were then either left uninduced or induced with IPTG (0.2 mM) and grown for a further 1 hour. After harvesting by centrifugation (4,000 rpm, 10 min, in Falcon tubes using a benchtop instrument), the cells were washed three times with 40 ml transport buffer (150 mM KCl, 5 mM MES, pH 6.6) and then resuspended in the same buffer to an A680 of 2.0. The basic method for the assay is described as follows. Cells were energised for building up the proton gradient to drive substrate transport by incubating aliquots of the suspension (955 μl) with 20 mM glycerol (20 μl of 1 M) and with bubbled air in a bijou bottle held in a water jacket at 25 °C. After exactly 3 minutes, radiolabelled substrate at a concentration of 50 μM (25 μl of a 2 mM stock solution) was added with brief mixing. At time points of 0.25, 1, 2, 5, 7.5 and 10 minutes after adding the radiolabelled substrate, 100 μl aliquots were transferred to cellulose nitrate filters (0.45 μm pore size), pre-soaked in transport buffer, on a vacuum manifold and washed immediately with transport buffer (3 ml) three times. The filters were transferred to scintillation vials with 10 ml Emulsifier Safe liquid scintillation fluid (Perkin Elmer Ltd, UK) and incubated overnight. The level of radioactivity retained by the cells was measured by liquid scintillation counting (Packard Tri-Carb 2100TR instrument). Background counts were measured from washing filters under vacuum in the absence of cells or radiolabelled substrate. Standard counts were measured by transferring 4 μl of the radiolabelled substrate stock solution (containing 8 nmol) directly to a washed filter in the vial. The uptake of radiolabelled substrate into the cells was calculated using the following equation: Uptake (nmol/mg cells) = (Cell counts – Background counts) x (Total assay volume/Sample taken volume) x (1/mg of cells) x (Moles of standard/Standard counts); where dry weight of cells (mg) = Total assay volume (ml) x A680 x 0.68. The times of sampling and/or of added radiolabelled substrate concentration were varied for kinetic analyses. To test the effect of potential competing compounds on 3H-cytosine uptake the unlabelled compound was added from a stock solution (50 mM) in 100% DMSO to the cells prior to the energisation period, the final concentration of the unlabelled compound was 500 M and the final concentration of DMSO was 1%. Relative uptake values were measured as a percentage of those obtained from samples in the absence of any added unlabelled compound or DMSO.
8.2.5. Protein solubilisation and purification
Inner membranes were solubilised for up to 4 hours at 4 °C in solubilisation buffer [20% (v/v) glycerol, 300 mM NaCl, 1% (w/v) SDS, 20 mM imidazole, 20 mM Tris-HCl (pH 8.0)]. The membranes were then sedimented at 131,000 x g for 1 hour at 4 °C to remove the insoluble fraction. For purification by immobilisedmetal affinity chromatography (IMAC), the supernatant was incubated with NiNTA resin for 2 hours at 4 °C with mixing and then transferred to a BioRad column and the supernatant was run through the column to elute unbound components. The resin was washed with 150-200 ml of wash buffer 1 [10% (v/v) glycerol, 0.05% (w/v) SDS, 20 mM imidazole, 10 mM Tris-HCl (pH 8.0)] to remove any remaining unbound material. The His6-tagged protein was removed by addition of elution buffer [2.5% (v/v) glycerol, 0.05% (w/v) SDS, 200 mM imidazole, 10 mM Tris-HCl (pH 8.0)]. Volumes of eluted samples were reduced to 3 ml using Vivaspin 20 tube concentrators (4,000 xg) with molecular weight cut off (MWCO) 100 kDa. The 3 ml sample was then applied to a BioRadEconopac 10 DG desalting column to remove the high concentration of imidazole. When the sample had run into the column, 4.5 ml of wash buffer 2 [2.5% (v/v) glycerol, 0.05% (w/v) SDS, 10 mM Tris-HCl (pH 8.0)] was applied and the eluted fraction was collected in a Vivaspin 6 tube (MWCO 100 kDa) and spun at 4,000 x g. Purified protein concentrated to 5-30 mg/ml was dispensed into aliquots, flash frozen in liquid nitrogen and stored at -80 °C.
8.2.6. Circular dichroism spectroscopy
The secondary structure content of purified protein was measured by far-UV circular dichroism (CD) spectroscopy using a CHIRASCAN instrument (Applied Photophysics, UK) at 20 ºC with constant nitrogen flushing. Protein samples (0.15 mg/ml) in 10 mM NaPi (pH 7.5) buffer plus 0.05% DDM were analysed in a Hellma quartz cuvette with a 1.0 mm pathlength. Measurements in the wavelength range 180-260 nm used a scan rate of 1 nm/second. A spectrum of buffer alone was subtracted from all sample spectra. Thermal stability was analysed by ramping the temperature from 5-90 ºC and finally back to 5 ºC, where each increment was held for 60 seconds before a measurement was made. Changes in secondary structure were monitored at 209 nm. Melting temperatures were estimated using Global Analysis CD software-3. CD ellipticity values were converted to mean residue ellipiticity (MRE, deg.cm-2.dmol-1).
8.3. RESULTS AND DISCUSSION
8.3.1. Database and computational analysis of V. parahaemolyticus protein VPA1242
According to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) (Kanehisa et al., 2016), the gene for V. parahaemolyticus protein VPA1242 (UniProt entry Q87GS3, 412 residues) is predicted to code for a cytosine permease and is immediately upstream from the gene that codes for protein VPA1243, which is predicted to be a cytosine deaminase. Indeed, VPA1242 shares 75.2% sequence identity with E. coli cytosine permease CodB and the E. coli cytosine-inducible operon codBA encodes CodB (UniProt entry P0AA82) followed by a cytosine deaminase CodA (UniProt entry P25524) (Danielsen et al., 1992; Qi and Turnbough, 1995). E. coli can utilise cytosine as its sole source of nitrogen by using CodB to mediate uptake of exogenous cytosine and CodA to catalyse hydrolytic deamination of cytosine to uracil (a source of pyrimidines) and ammonia (a source of nitrogen) (Danielsen et al., 1992). Based on separate sequence alignments with characterised bacterial NCS1 family proteins, VPA1242 shares overall sequence homologies of 47.3% (22.8% identical, 24.5% highly similar) with Mhp1, 45.1% (24.8% identical, 20.3% highly similar) with PucI and 89.5% (75.2% identical, 14.3% highly similar) with CodB. This further confirms that VPA1242 is most closely related with E. coli CodB and suggests that VPA1242 is a transporter of cytosine.
Analysis of the VPA1242 amino acid sequence using the prediction tools TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) (Krogh et al., 2001) and TOPCONS (http://topcons.cbr.su.se/) (Bernsel et al., 2009) suggested that protein contains eleven or twelve transmembrane-spanning -helices, respectively (Figure 1). Based on structurally characterised NCS1 family proteins and a demonstrated reliability for TOPCONS (Hennerdal and Elofsson, 2011; Tsirigos et al., 2015; Saidijam et al., 2018), twelve transmembrane helices is most likely to be correct. In this case, both the N- and C-terminal ends of the protein are predicted to reside at the cytoplasmic side of the membrane (Figure 2). This is the same as the membrane topology predicted for E. coli CodB (Danielsen et al., 1995). Locations of the twelve transmembrane helices predicted by TOPCONS in the sequence of VPA1242 align relatively well with those of transmembrane regions in the Mhp1 sequence based on its crystal structure (Figure 3).
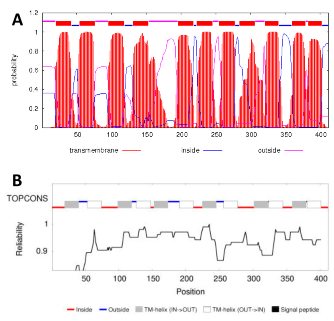
Figure 1. Predictions of transmembrane helices in V. parahaemolyticus protein VPA1242. The amino acid sequence of VPA1242 was taken from the UniProt KnowledgeBase (http://www.uniprot.org/) and analysed by membrane topology prediction tools TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) (Krogh et al., 2001) (A) and TOPCONS (http://topcons.cbr.su.se/) (Bernsel et al., 2009) (B).
Figure 2. Predicted membrane topology of V. parahaemolyticus transport protein VPA1242. Diagram for the putative membrane topology of the VPA1242 transport protein of V. parahaemolyticus based on TOPCONS prediction for transmembrane helices (Figure 1). Twelve transmembrane-spanning -helices are predicted and residues are coloured to show those that are identical (red) or highly similar (blue) compared with corresponding positions in the sodium and substrate binding site of Mhp1.
Based on the nine residues involved in substrate binding in Mhp1 (Simmons et al., 2014), we can compare the residues at corresponding positions in other characterised bacterial NCS1 proteins to look for possible explanations about the origin of substrate specificity (Table 1 and Figure 3). Out of these residues, only three VPA1242 residues (Ala35, Trp108, Leu325) are identically conserved with those in Mhp1, whilst all nine residues are identically conserved in VPA1242 and CodB. In comparison, five out of nine residues are identically conserved in PucI and Mhp1. Only two out of nine VPA1242/CodB residues (Trp108, Leu325) are identically conserved with those in PucI. These differences appear to account for the significantly different substrate specificities of VPA1242 and CodB compared with Mhp1and PucI. These observations further confirm the close relationship between VPA1242 and CodB. Cautions should of course be made about making possible structural explanations for differences in substrate specificity between NCS1 proteins, as demonstrated by studies on the fungal transporters (Hamari et al., 2009; Krypotou et al. 2015).

Figure 3. Conservation of residues between V. parahaemolyticus protein VPA1242, Mhp1 and CodB. Amino acid sequences of VPA1242 from V. parahaemolyticus (Q87GS3), Mhp1 from M. liquefaciens (D6R8X8) and CodB from E. coli (P0AA82) were taken from the UniProt KnowledgeBase (http://www.uniprot.org/) and aligned using Clustal Omega
(http://www.ebi.ac.uk/Tools/msa/clustalo/) (Sievers et al., 2011). Conserved residues are indicated below the sequences as identical (*), highly similar (:) and similar (.). Residues in the substrate binding site of Mhp1 are highlighted (green) and residues at these positions are coloured for those that are identical (red) or highly similar (blue) in the three proteins. Helical regions in Mhp1 based on the crystal structure of Mhp1 with bound benzylhydantoin (PDB 4D1B) (Simmons et al., 2014) are highlighted: transmembrane helix (grey), internal helix (cyan), external helix (pink). Putative transmembrane helices in VPA1242 based on TOPCONS prediction are also highlighted (grey).
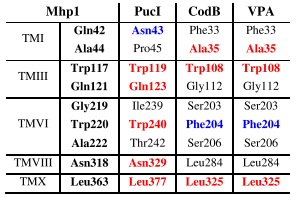
Table 1. Conservation of substrate binding residues in characterised bacterial NCS1 family proteins. Residues in the substrate binding site of crystallographically defined Mhp1 (5-arylhydantoins) are compared with those at the corresponding positions in PucI (allantoin), CodB (cytosine) and VPA1242 (cytosine) based on sequence alignments. Residues are coloured to indicate those that are identically conserved (red) or highly similar (blue) compared with residues in Mhp1.
8.3.2. Gene cloning and amplified expression of V. parahaemolyticus protein VPA1242 in E. coli
V. parahaemolyticus protein VPA1242 was tractable to an established strategy for amplified expression in E. coli of bacterial NCS1 family proteins using plasmid pTTQ18 with concomitant introduction of a C-terminal His6 tag to assist protein purification (Ward et al., 1999; Saidijam et al., 2003; Saidijam et al., 2005; Clough et al., 2006; Suzuki and Henderson, 2006; Szakonyi et al., 2007; Gordon et al., 2008; Ma et al., 2008, 2013; Bettaney et al., 2013; Ma et al., 2016). Criteria included availability of genomic DNA for the organism of origin, absence of internal EcoRI and PstI restriction sites in the gene for the protein of interest, a predicted cytoplasmic location of the protein C-terminus and availability of radiolabelled potential substrates for transport activity assays.
The V. parahaemo