colliding with a single H 2 molecule. The probability for such a collision should be proportional to






. However, experimentally we observe (see Table 6.4) that the rate law for this reaction is ()
As a second example, consider the reaction
()
It would seem reasonable to assume that this reaction occurs as a single collision in which an
oxygen atom is exchanged between the two molecules. However, the experimentally observed rate
law for this reaction is
()
In this case, the [ CO] concentration does not affect the rate of the reaction at all, and the
concentration is squared. These examples demonstrate that the rate law for a reaction cannot be
predicted from the stoichiometric coefficients and therefore that the collision model does not
account for the rate of the reaction. There must be something seriously incomplete with the
collision model.
The key assumption of the collision model is that the reaction occurs by a single collision. Since
this assumption leads to incorrect predictions of rate laws in some cases, the assumption must be
invalid in at least those cases. It may well be that reactions require more than a single collision to
occur, even in reactions involving just two types of molecules as in Equation. Moreover, if more than two molecules are involved as in Equation, the chance of a single collision involving all of the reactive molecules becomes very small. We conclude that many reactions, including those in
Equation and Equation, must occur as a result of several collisions occurring in sequence, rather than a single collision. The rate of the chemical reaction must be determined by the rates of the
individual steps in the reaction.
Each step in a complex reaction is a single collision, often referred to as an elementary process.
In single collision process step, our collision model should correctly predict the rate of that step.
The sequence of such elementary processes leading to the overall reaction is referred to as the
reaction mechanism. Determining the mechanism for a reaction can require gaining substantially
more information than simply the rate data we have considered here. However, we can gain some
progress just from the rate law.
Consider for example the reaction in Equation described by the rate law in Equation. Since the rate law involved
, one step in the reaction mechanism must involve the collision of two NO 2
molecules. Furthermore, this step must determine the rate of the overall reaction. Why would that
be? In any multi-step process, if one step is considerably slower than all of the other steps, the rate
of the multi-step process is determined entirely by that slowest step, because the overall process
cannot go any faster than the slowest step. It does not matter how rapidly the rapid steps occur.
Therefore, the slowest step in a multi-step process is thus called the rate determining or rate
limiting step.

This argument suggests that the reaction in Equation proceeds via a slow step in which two NO 2
molecules collide, followed by at least one other rapid step leading to the products. A possible
mechanism is therefore
(6.1)
Step 1
NO 2+ NO 2→ NO 3+ NO
(6.2)
Step 2
NO 3+ CO→ NO 2+ CO 2
If Step 1 is much slower than Step 2, the rate of the reaction is entirely determined by the rate of
Step 1. From our collision model, the rate law for Step 1 must be
, which is consistent
with the experimentally observed rate law for the overall reaction. This suggests that the
mechanism in Equation 6.1 and Equation 6.2 is the correct description of the reaction process for
Equation, with the first step as the rate determining step.
There are a few important notes about the mechanism. First, one product of the reaction is
produced in the first step, and the other is produced in the second step. Therefore, the mechanism
does lead to the overall reaction, consuming the correct amount of reactant and producing the
correct amount of reactant. Second, the first reaction produces a new molecule, NO 3, which is
neither a reactant nor a product. The second step then consumes that molecule, and NO 3 therefore
does not appear in the overall reaction, Equation. As such, NO 3 is called a reaction intermediate.
Intermediates play important roles in the rates of many reactions.
If the first step in a mechanism is rate determining as in this case, it is easy to find the rate law for
the overall expression from the mechanism. If the second step or later steps are rate determining,
determining the rate law is slightly more involved. The process for finding the rate law in such a
case is illustrated in Exercise 11..
Review and Discussion Questions
Exercise 1.
When C 60 O 3 in toluene solution decomposes, O 2 is released leaving C 60 O 3 in solution.
Based on the data in Figure 6.2 and Figure 6.3, plot the concentration of C 60 O as a function of time.
How would you define the rate of the reaction in terms of the slope of the graph from above? How is the rate of appearance of C 60 O related to the rate of disappearance of C 60 O 3? Based on this, plot the rate of appearance of C 60 O as a function of time.








Exercise 2.
The reaction
was found in this study to have rate law given by
with k=0.070 s-1.
How is the rate of appearance of NO 2 related to the rate of disappearance of N 2 O 5? Which rate is larger?
Based on the rate law and rate constant, sketch a plot of
,
, and
versus time all on the
same graph.
Exercise 3.
For which of the reactions listed in Table 6.4 can you be certain that the reaction does not occur as a single step collision? Explain your reasoning.
Exercise 4.
Consider two decomposition reactions for two hypothetical materials, A and B. The
decomposition of A is found to be first order, and the decomposition of B is found to be second
order.
Assuming that the two reactions have the same rate constant at the same temperature, sketch [ A]
and [ B] versus time on the same graph for the same initial conditions, i.e. [ A]0=[ B]0.
Compare the half-lives of the two reactions. Under what conditions will the half-life of B be less
than the half-life of A? Under what conditions will the half-life of B be greater than the half-life
of A?
Exercise 5.
A graph of the logarithm of the equilibrium constant for a reaction versus is linear but can have
either a negative slope or a positive slope, depending on the reaction, as was observed here.
However, the graph of the logarithm of the rate constant for a reaction versus has a negative
slope for essentially every reaction. Using equilibrium arguments, explain why the graph for the
rate constant must have a negative slope.
Exercise 6.
Using Equation and the data in Table 6.5, determine the activation energy for the reaction
.







Exercise 7.
We found that the rate law for the reaction
is
. Therefore, the reaction
is second order overall but first order in H 2. Imagine that we start with
and we measure
versus time. Will a graph of
versus time be linear or will a graph of
versus time be
linear? Explain your reasoning.
Exercise 8.
As a rough estimate, chemists often assume a rule of thumb that the rate of any reaction will
double when the temperature is increased by 10 °C.
What does this suggest about the activation energies of reactions?
Using Equation, calculate the activation energy of a reaction whose rate doubles when the temperature is raised from 25 °C to 35 °C.
Does this rule of thumb estimate depend on the temperature range? To find out, calculate the
factor by which the rate constant increases when the temperature is raised from 100 °C to 110 °C, assuming the same activation energy you found above. Does the rate double in this case?
Exercise 9.
Consider a very simple hypothetical reaction A+ B↔2 C which comes to equilibrium.
At equilibrium, what must be the relationship between the rate of the forward reaction, A+ B→2 C
and the reverse reaction 2 C→ A+ B ?
Assume that both the forward and reverse reactions are elementary processes occurring by a single
collision. What is the rate law for the forward reaction? What is the rate law for the reverse
reaction?
Using the previous results from here and here, show that the equilibrium constant for this reaction can be calculated from
, where kf is the rate constant for the forward reaction and kr
is the rate constant for the reverse reaction.
Exercise 10.
Consider a very simple hypothetical reaction A+ B↔ C+ D . By examining Figure 6.8, provide and explain the relationship between the activation energy in the forward direction, Ea, f, and in the


reverse direction, Ea, r . Does this relationship depend on whether the reaction is endothermic (Figure 6.8a) or exothermic (Figure 6.8b)? Explain.
Exercise 11.
For the reaction
, the rate law is
. Although this suggests that the
reaction is a one-step elementary process, there is evidence that the reaction occurs in two steps,
and the second step is the rate determining step:
(6.3)
Step 1
I 2↔2 I
(6.4)
Step 2
H 2+2 I→2 HI
Where Step 1 is fast and Step2 is slow.
If the both the forward and reverse reactions in Step 1 are much faster than Step2, explain why
Step 1 can be considered to be at equilibrium.
What is the rate law for the rate determining step?
Since the rate law above depends on the concentration of an intermediate I, we need to find that intermediate. Calculate [ I] from Step 1, assuming that Step1 is at equilibrium.
Substitute [ I] from above into the rate law found previously to find the overall rate law for the reaction. Is this result consistent with the experimental observation?
Solutions



Chapter 7. Equilibrium and the Second Law of
Thermodynamics
Foundation
We have observed and defined phase transitions and phase equilibrium. We have also observed
equilibrium in a variety of reaction systems. We will assume an understanding of the postulates of
the Kinetic Molecular Theory and of the energetics of chemical reactions.
Goals
We have developed an understanding of the concept of equilibrium, both for phase equilibrium
and reaction equilibrium. As an illustration, at normal atmospheric pressure, we expect to find
H 2 O in solid form below 0°C, in liquid form below 100°C, and in gaseous form above 100°C.
What changes as we move from low temperature to high temperature cause these transitions in
which phase is observed? Viewed differently, if a sample of gaseous water at 120°C is cooled to
below 100°C, virtually all of the water vapor spontaneously condenses to form the liquid:
By contrast, very little of liquid water at 80°C spontaneously
converts to gaseous water:
We can thus rephrase our question as,
what determines which processes are spontaneous and which are not? What factors determine
what phase is "stable"?
As we know, at certain temperatures and pressures, more than one phase can be stable. For
example, at 1 atm pressure and 0°C,
Small variations in the amount of
heat applied or extracted to the liquid-solid equilibrium cause shifts towards liquid or solid
without changing the temperature of the two phases at equilibrium. Therefore, when the two
phases are at equilibrium, neither direction of the phase transition is spontaneous at 0°C. We
therefore need to understand what factors determine when two or more phases can co-exist at
equilibrium.
This analysis leaves unanswered a series of questions regarding the differences between liquids
and gases. The concept of a gas phase or a liquid phase is not a characteristic of an individual
molecule. In fact, it does not make any sense to refer to the "phase" of an individual molecule. The
phase is a collective property of large numbers of molecules. Although we can discuss the
importance of molecular properties regarding liquid and gas phases, we have not discussed the
factors which determine whether the gas phase or the liquid phase is most stable at a given
temperature and pressure.
These same questions can be applied to reaction equilibrium. When a mixture of reactants and
products is not at equilibrium, the reaction will occur spontaneously in one direction or the other
until the reaction achieves equilibrium. What determines the direction of spontaneity? What is the
driving force towards equilibrium? How does the system know that equilibrium has been
achieved? Our goal will be to understand the driving forces behind spontaneous processes and the
determination of the equilibrium point, both for phase equilibrium and reaction equilibrium.
Observation 1: Spontaneous Mixing
We begin by examining common characteristics of spontaneous processes, and for simplicity, we
focus on processes not involving phase transitions or chemical reactions. A very clear example of
such a process is mixing. Imagine putting a drop of blue ink in a glass of water. At first, the blue
dye in the ink is highly concentrated. Therefore, the molecules of the dye are closely congregated.
Slowly but steadily, the dye begins to diffuse throughout the entire glass of water, so that
eventually the water appears as a uniform blue color. This occurs more readily with agitation or
stirring but occurs spontaneously even without such effort. Careful measurements show that this
process occurs without a change in temperature, so there is no energy input or released during the
mixing.
We conclude that, although there is no energetic advantage to the dye molecules dispersing
themselves, they do so spontaneously. Furthermore, this process is irreversible in the sense that,
without considerable effort on our part, the dye molecules will never return to form a single
localized drop. We now seek an understanding of how and why this mixing occurs.
Consider the following rather abstract model for the dye molecules in the water. For the glass, we
take a row of ten small boxes, each one of which represents a possible location for a molecule,
either of water or of dye. For the molecules, we take marbles, clear for water and blue for ink.
Each box will accommodate only a single marble, since two molecules cannot be in the same
place as the same time. Since we see a drop of dye when the molecules are congregated, we model
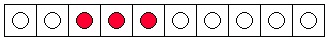
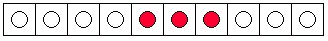
a "drop" as three blue marbles in consecutive boxes. Notice that there are only eight ways to have
a "drop" of dye, assuming that the three dye "molecules" are indistinguishable from one another.
Two possibilities are shown in Figure 7.1a and Figure 7.1b. It is not difficult to find the other six.
(a) An unmixed state.
(b) Another unmixed state.
(c) A mixed state.

(d) Another mixed state.
Figure 7.1. Arrangement of Three Ink Molecules
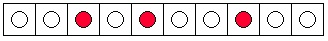
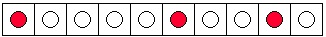
By contrast, there are many more ways to arrange the dye molecules so that they do not form a
drop, i.e. , so that the three molecules are not together. Two possibilities are shown in Figure 7.1c
and Figure 7.1d. The total number of such possibilities is 112. (The total number of all possible arrangements can be calculated as follows: there are 10 possible locations for the first blue
marble, 9 for the second, and 8 for the third. This gives 720 possible arrangements, but many of
these are identical, since the marbles are indistinguishable. The number of duplicates for each
arrangement is 6, calculated from three choices for the first marble, two for the second, and one
for the third. The total number of non-identical arrangements of the molecules is
.) We
conclude that, if we randomly place the 3 marbles in the tray of 10 boxes, the chances are only 8
out of 120 (or 1 out of 15) of observing a drop of ink.
Now, in a real experiment, there are many, many times more ink molecules and many, many times
more possible positions for each molecule. To see how this comes into play, consider a row of 500
boxes and 5 blue marbles. (The mole fraction of ink is thus 0.01.) The total number of distinct
configurations of the blue marbles in these boxes is approximately 2×1011. The number of these
configurations which have all five ink marbles together in a drop is 496. If the arrangements are
sampled randomly, the chances of observing a drop of ink with all five molecules together are
thus about one in 500 million. The possibilities are remote even for observing a partial "droplet"
consisting of fewer than all five dye molecules. The chance for four of the molecules to be found
together is about one in 800,000. Even if we define a droplet to be only three molecules together,
the chances of observing one are less than one in 1600.
We could, with some difficulty, calculate the probability for observing a drop of ink when there
are 1023 molecules. However, it is reasonably deduced from our small calculations that the
probability is essentially zero for the ink molecules, randomly distributed into the water
molecules, to be found together. We conclude from this that the reason why we observe ink to
disperse in water is that the probability is infinitesimally small for randomly distributed dye
molecules to be congregated in a drop.
Interestingly, however, when we set up the real ink and water experiment, we did not randomly
distribute the ink molecules. Rather, we began initially with a drop of ink in which the dye
molecules were already congregated. We know that, according to our kinetic theory, the molecules
are in constant random motion. Therefore, they must be constantly rearranging themselves. Since
these random motions do not energetically favor any one arrangement over any other one
arrangement, we can assume that all possible arrangements are equally probable. Since most of
the arrangements do not correspond to a drop of ink, then most of the time we will not observe a
drop. In the case above with five blue marbles in 500 boxes, we expect to see a drop only once in
every 500 million times we look at the "glass". In a real glass of water with a real drop of ink, the chances are very much smaller than this.
