MANIFESTATIONS OF GENETIC DISEASES
1.Adiposity, hyperthermia, oligomenorrhea and parotid swelling syndrome :
This is also known as AHOP or AOP syndrome. It is caused by a hereditary alteration of some part of the diencephalon. It is thought to be transmitted as sex linked dominant character. Clinical features :
The clinical findings are usually limited to females. It consists of adiposity, hyperthermia, oligomenorrhea, parotid swelling and psychic disturbances. The parotid swelling usually begins at the time of puberty and is bilateral. Occasionally the submandibular glands are also involved.
2.AARSKOG SYNDROME
(Faciodigitogenital Syndrome)
Aarskog–Scott syndrome is a rare disease inherited as autosomal dominant or X-linked and characterized by short stature, facial abnormalities, skeletal and genital anomalies. The Aarskog– Scott syndrome (AAS) is also known as the Aarskog syndrome, faciodigitogenital syndrome, shawl scrotum syndrome and faciogenital dysplasia.
Clinical Features:
The Aarskog–Scott syndrome is a disorder with short stature, hypertelorism, downslanting palpebral fissures, anteverted nostrils, joint laxity, shawl scrotum, and mental retardation. The physical phenotype varies with age and postpuberal males may have only minor remnant manifestations of the prepuberal phenotype.
Growth mild to moderate short stature evident by 1–3 years of age
3) Adrenogenital syndrome :
4) Aglossia adactylia syndrome :
(Hypoglossia hypodactylia syndrome)
The etiology for the condition is unknown. There is no evidence that heredity plays a role in its genesis. There is no sex predilection. There is absence of tongue associated with failure of development of digits. The tongue in most cases is completely absent, or may be present as a small lobule in posterior part of the mouth. But the speech is not severely impaired. The sublingual muscular ridges may be markedly enlarged. Sublingual and submandibular glands may be hypertrophic. The mandible is usually small and poorly developed. Lower incisors have been noted to be missing. Extremities are severely affected, from peromelia to absence of single digit. Both hands and feet are usually involved. Syndactyly has also been reported. Intelligence in these patients is not impaired.
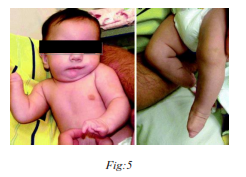
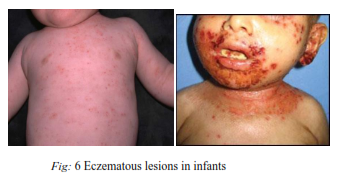
5) Aldrich’s syndrome :
(‘Wiskot Aldrich’ syndrome)
Wiskott–Aldrich syndrome was linked in 1994[6] to mutations in a gene on the short arm of the X chromosome, which was termed Wiskott-Aldrich syndrome protein gene (WASp). The disease X-linked thrombocytopenia was later discovered to be also due to WASp mutations, but different ones from those that cause full-blown Wiskott–Aldrich syndrome
Classification:
Jin et al. (2004) employ a numerical grading of severity:
0.5: intermittent thrombocytopenia
1.0: thrombocytopenia and small platelets (microthrombocytopenia)
2.0: microthrombocytopenia plus normally responsive eczema or occasional upper respiratory tract infections
2.5: microthrombocytopenia plus therapy-responsive but severe eczema or airway infections requiring antibiotics
3.0: microthrombocytopenia plus both eczema and airway infections requiring antibiotics
4.0: microthrombocytopenia plus eczema continuously requiring therapy and/or severe or life-threatening infections
5.0: microthrombocytopenia plus autoimmune disease or malignancy
6) Amelo-onycho-hypohydrotic syndrome :
A rare disorder characterized primarily by tooth and nail abnormalities and reduced sweating ability Clincal features:
7) Antley-Bixler Syndrome:
(Trapezoidocephaly-synostosis syndrome)
It is a rare, very severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.
The disorder is suggested to be genetically heterogenous as two distinct gene mutation (FGFR2 & POR gene) are responsible for phenotypic expression of this syndrome.
General manifestations:
a) Coronal and lambdoid craniosynostosis
b) Brachycephaly
c) Proptosis
d) Choanal stenosis/atresia
e) Maxillary hypoplasia
f) Humeroraadial synoostosis
g) Camptodactyly (fused interphalangeal joints in the fingers)
h) Multiple contractures
i) Cardiac malformations
j) Nasal/anal atresia
8) Albright’s Syndrome :
(Polyostotic fibrous dysplasia, cutaneous pigmentation and endocrine disorders, Brown spot syndrome).
Etiology of the syndrome is obscure. Heredity does not appear to be a factor in the syndrome. The syndrome consists of a classic triad of polyostotic fibrous dysplasia, pigmentation of skin and rarely mucous membrane and endocrine dysfunction.
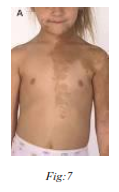
Oral manifestations –
Mandible is more commonly involved. It is enlarged, expanded and distorted. Pigmentation of the lips and buccal mucosa is rarely seen.
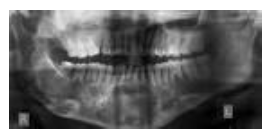
Radiographic appearance :
Medullary portions of bone are rarefied and present irregular trabeculations. Radiographically the condition presents as a unilocular, multilocular or ground-glass or peau de orange appearance.
9. Anderson Syndrome:
(Familial Osteodysplasia)
It is inherited as an autosomal recessive trait. It is characterized by craniofacial and skeletal anomalies, presence of hyperuricemia and diastolic hypertension.
Clincal features :
1. Mandibular prognathism.
2. Hypoplastic.
3. Maxilla and resultant malocclusion.
4. The mandible is with wide angle, increased body length, and reduced ramus and body height.
5. Spine abnormalities
6. Collar bone abnormalities
7. Pelvic abnormalities
8. Thigh bone abnormalities
9. Foot abnormalities
10. Skull abnormalities
11. High uric acid level in blood
12. Diastolic hypertension
13. Recurring fractures
14. Abnormal tooth positioning
15. Flat nose
16. Flat cheek bones
17. Kyphosis
18. Large nose
19. Pointy chin
20. Dental decay
21. Curved fifth finger
22. High blood pressure
23. Thick eyebrows
10) Angioosteohypertrophy Syndrome :
(Klippel-Trenaunay-Weber syndrome, Hemangiectatic hypertrophy)
The syndrome consists of unilateral nevus flammeus of the skin arranged segmentally, varices and hypertrophy of the skeletal and soft tissue developing at about the same time. It is transmitted as either an autosomal dominant or recessive trait.
Oral manifestations :
The soft and hard palate are most frequently involved. Angiomatosis of the tongue and pharynx were also reported. There is asymmetry of face because of bony hypertrophy; resulting in malocclusion and premature eruption of teeth on the involved side.
11) Ankyloglossum Superius Syndrome :
It consists of congenital ankylosis of tongue to the hard palate and anomalies of hands and feet such as hypoplasia or aplasia of digits and syndactyly.
Oral manifestations :
The tongue is usually attached to the hard palate, but may also be attached to upper alveolar ridge. The tongue is attached usually in the anterior part. The tongue functions like mobility and extrusion beyond the teeth are hindered. The upper central portion of lip is usually hypoplastic and mandible has been described as hypoplastic. Cleft lip and palate has also been recorded.
12) Aortic arch syndrome :
(Subclavian artery occlusive syndrome, subclavian steal syndrome, vetebral –basilar artery occlusive syndrome, pulseless diesease)
Aortic arch syndrome problems are most often associated with trauma, blood clots, or malformations that develop before birth. The arteries' defects result in abnormal blood flow to the head, neck, or arms. In children, there are multiple types of aortic arch syndromes, including:
Symptoms vary according to the affected artery, but may include:
13) Apert’s Syndrome (Acrocephalosyndactyly) :
It is classified as a branchial arch syndrome, affecting thefirst branchial (or pharyngeal) arch, the precursor of the maxilla andmandible.

Apert syndrome is a developmental malformation characterized by–
1) Craniosynostosis
2) A cone shaped calvarium
3) Midface hypoplasia
4) Pharyngeal attenuation
5) Ocular manifestations
6) Syndactyly of the hands and feet
7) Mutations of either Ser252Trp or Pro253Arg in fibroblast growth factor receptor 2 (FGFR2) are responsible for nearly all known cases of Apert syndrome.
8) Common dental features of acrocephalosyndactyly are a higharched palate, pseudomandibular prognathism (appearing as mandibular prognathism), a narrow palate, and crowding of the teeth.
Clinical features :
a) Face :
Face is usually asymmetric. The middle third of the face is underdeveloped and flat, producing a relative prognathism. The nose is usually described as parrot’s beak. Hypertelorism, exophthalmos and strabismus are often reported.
b) Skull :
The cranium is usually oxycephalic in appearance. The apex of the cranium is located near to or anterior to the bregma. Anterior fontanelle is open in numerous patients.
14) Ascher’s Syndrome:
(Laffer-Ascher Syndrome)
The etiology is unknown. However hormonal dysfunction, trauma and heredity are suggested as etiological agents. It is characterized by repeated episodes of lip and eyelid edema and occasionally euthyroid goiter. The syndrome generally occurs within the first 20 years of life
Oral manifestations :
There is a horizontal line running between the inner and outer parts of upper lip. Very rarely lower lip is also enlarged. The enlargement of lip may exist from childhood. The ‘extra lip’ or the tissue is usually seen when the patient smiles or during talking.
Microscopic examination of this excessive tissue usually consists of loose areolar tissue and hyperplastic mucous glands.
15) Ataxia Telangiectasia Syndrome:
Also referred to as Louis–Bar syndrome is a rare, neurodegenerative, inherited disease causing severe disability. Ataxia refers to poor coordination and telangiectasia to small dilated blood vessels, both of which are hallmarks of the disease. Mutations in the ATM gene cause ataxia-telangiectasia. The ATM gene provides instructions for making a protein that helps control cell division and is involved in DNA repair.
It is a neurocutaneous disorder transmitted as an autosomal recessive trait.
Clinical features:
a) Face:
Face is relaxed, dull or sad. Shoulders are drooped and the head is tilted to one side. There are mild athetoid movements around the shoulders.
b) Oral manifestations:
Telangiectasia of the hard and soft palate has been reported. There is drooling of saliva and speech is slow, slurred and interrupted. Eye movements are slow and halted during lateral and upward gaze.
Warning Signs of a Swallowing Problem
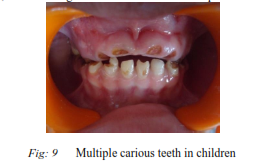
16) Baby Bottle Syndrome :
Bottle mouth syndrome; also called as Nursing bottle caries. This syndrome has been attributed to prolonged use of:
1. Nursing bottle containing milk or milk formula, fruit juice or sweet water.
2. Breastfeeding or
3. Sugar or honey, sweetened pacifiers.
Clinical features :
It presents as a widespread carious destruction of deciduous teeth, most commonly the maxillary incisors, followed by first molars and then the cuspids. But the mandibular incisors are spared, which distinguishes this disease from rampant caries.
17) Beckwith-Wiedemann Syndrome :
Five common features used to define BWS are: macroglossia, macrosomia (birth weight and length greater than the 90th percentile), midline abdominal wall defects (omphalocele/ exomphalos, umbilical hernia, diastasis recti), ear creases or ear pits, and neonatal hypoglycemia (low blood sugar after birth).
Oral features:
1. Macroglossia
2. Malocclusion,
3. Anterior open bite
4. Widely spaced teeth likely due to macroglossia
5. Speech problems due to macroglossia
Most children with BWS do not have all of these five features. In addition, some children with BWS have other findings including: nevus flammeus, prominent occiput, midfacehypoplasia, hemihypertrophy, genitourinary anomalies (enlarged kidneys), cardiac anomalies, musculoskeletal abnormalities, and hearing loss. Also, some premature newborns with BWS do not have macroglossia until closer to their anticipated delivery date.
18) Beal syndrome:
(Congenital contractual arachnodactyly)
It is a rare congenital connective tissue disorder. It is caused by a mutation in FBN2 gene on chromosome 5q23. Contractures of varying degrees at birth, mainly involving the large joints, are present in all affected children. Elbows, knees and fingers are most commonly involved. The contractures may be mild and tend to reduce in severity.
Manifestations:
a. Cranial deformity
b. Bowed long bones
c. Muscle hypoplasia
d. Mitral valve prolapse
e. Arthrogyposis
f. Arachnodactyly
g. Kyphoscoliosis
h. Abnormal helix of ear (crumpled ear)
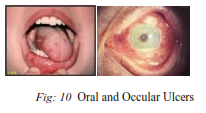
19) Behcet’s Syndrome:
Behcet’s disease is a systemic vasculitis characterized by recurrent oral and genital ulcers, and ocular inflammation, and which may involve the joints, skin, central nervous system and gastrointestinal tract. It is most common in those of Mediterranean and Eastern origin, although it also affects Caucasians. Genetic susceptibility :
The genetic locus most widely studied in Behcet’s disease is the human leukocyte antigen (HLA) complex on chromosome.
Clinical features :
1. Recurrent aphthous ulceration –
2. Skin disease –
20) Blepharonasal Facial Syndrome:
It is inherited as an autosomal dominant trait. It is characterized by joint disorders, craniofacial anomalies and mental retardation. Individual presents with microcephaly, and amtomongoloid slant of palpebral fissures, hypoplastic maxial, protruding lip and mild malocclusion as a result of mid facial hypoplasia.
21) Bloch-Sulzberger Syndrome:
Incontinentia pigmenti also known as "Bloch–Siemens syndrome, "Bloch–Sulzbergerdisease, "Bloch–Sulzberger syndrome "melanoblastosis cutis," and "naevus pigmentosus systematicus". It is a genetic disorder that affects the skin, hair, teeth, nails, and central nervous system. It is named due to its microscopic appearance.
It is transmitted as a sex-linked dominant trait, and it is lethal in males. More than 95% of reported cases occur in females. The etiology is not known. It is postulated that viral or infectious causes during pregnancy are predisposing factors.
a) Oral manifestations :
Oral changes are limited to the teeth. They include delayed teeth eruption, pegged or conical shaped crowns of teeth, missing teeth, malformed teeth and additional cusps. Both deciduous and permanent teeth are affected.
b) Skin and skin appendages :
After 2–3 days of birth, linear or grouped vesicles containing a honey colored serum appear on extremities. By the end of the first month the vesicles may disappear, recur or be replaced by irregularly distributed violaceous papules and inflammatory lesions. Pigmented macules, brownish-gray in color and arranged in a reticulated pattern or in streaks, whorls or patches over trunk and extremities are seen at birth. This pigmentation begins to fade within a few years. It is heavy melanin pigmentation of the epithelium, dropping down into clusters of chromatophores in the upper dermis (incontinence, which gives the disease its name and is considered the hallmark of the syndrome). There is generalized baldness. Rarely finger nails are dystrophic and the breasts asymmetric. During the vesicular stage, microscopic examination reveals dense intraepithelial (or occasionally subepithelial) vesicles containing eosinophils.
22) Bogorad’s Syndrome
(Crocodile tears syndrome, Gustatory lacrimation)
In this condition the patient exhibits profuse lacrimation when food is eaten; particularly hot and spicy food. It generally follows facial paralysis; either of Bell’s palsy type or the result of herpes zoster, head injury or intracranial operative trauma.
Facial nerve has several components – motor, sensory and autonomic – parasympathetic component. The autonomic parasympathetic fi bers arise in superior salivatory nucleus provide secretory and vasodilatory function to submandibular sublingual glands, via chorda tympani and to lacrimal gland the nerve of Wrisberg. If a lesion occurs proximal to geniculate ganglion, then during regeneration, fibers destined to submandibular and sublingual can get interchanged with those destined for lacrimal gland. Thus when gustatory stimuli a