quaternions. Journal of the Optical Society of America, 4:629-642.
E. A. Coutsias and C. Seok and K. A. Dill. (2004). Using quaternions to calculate RMSD.
Journal of Computational Chemistry, 25, 1849-1857.
Wallin, S., J. Farwer and U. Bastolla. (2003). Testing similarity measures with continuous
and discrete protein models . Proteins, 50:144-157.
References
1. Kabsch, W. (1976). A Solution for the Best Rotation to Relate Two Sets of Vectors. Acta
Crystallographica, 32, 922-923.
2. Kabsch, W. (1978). A Discussion of the Solution for the Best Rotation to Relate Two Sets of
Vectors. Acta Crystallographica, 34, 827-828.
3. Horn, Berthold K. P. (1986). Closed-form solution of absolute orientation using unit
quaternions. Journal of the Optical Society of America, 4, 629-642.
4. Coutsias, E. A., C. Seok and K. A. Dill. (1978). Using quaternions to calculate RMSD. Journal
4. Coutsias, E. A., C. Seok and K. A. Dill. (1978). Using quaternions to calculate RMSD. Journal
of Computational Chemistry, 25, 1849-1857.
5. Golub, G. H. and Loadn, C. F. V. (1996). Matrix Computations. (third). Johns Hopkins
University Press.
6. Wallin, S., J. Farwer and U. Bastolla. (2003). Testing similarity measures with continuous and
discrete protein models. Proteins, 50, 144-157.
7. Schwarzer, F. and Lotan, I. (2003). Approximation of protein structure for fast similarity
measures. ACM. Proceedings of the seventh annual international conference on research in
computational molecular biology.
Protein Classification, Local Alignment, and Motifs
Topics in this Module
Applications of Molecular Distance Measures
Local Matching: Geometric Hashing, Pose Clustering, and Match Augmentation
In a previous module, the topic of comparing and quantifying the distance between different conformations of a given molecule was explored. Structure-based comparison is also of interest
for distinct proteins, which lack the atom-by-atom correspondence necessary for RMSD
calculations. In this case, an alignment is performed either based on amino acid sequence or on
three-dimensional structure, and the subset of atoms successfully aligned are used as the basis for
calculating conformational distance. Computing distances among entire proteins by doing a global
alignment of their structures is useful for protein classification.
Protein Classification
Protein classification is motivated by the notion of "descriptive biology". When faced with
tremendous amounts of highly complex data, such as with the set of all proteins, one way to
understand the data is by classification: the act of associating or grouping proteins into classes
using certain criteria. One such criterion is protein sequence identity, where sequential similarity
led to the development of phylogenetic trees and multiple sequence analyses. The same is done in
protein structure classification, where the effort is to identify groups of similar proteins, with the
hope that this will yield information about their biochemical function and biological purpose.
Proteins are classified by simultaneously applying a number of criteria, including sequence
homology (evolutionary relatedness), function, folding motifs, structural features, and so on. The
resulting hierarchies and clusters of protein structures provide a notion of the distance between
two proteins and their structures. A couple of popular classification schemes are linked below.
Note that a fair amount of manual annotation and classification was necessary to build these
systems.
Protein Alignment
The core computational problem of protein classification, using sequence or structure, is the
problem of comparing two proteins. For structural classification, one method for comparison is
structural alignment, which identifies an ideal superimposition between two protein structures,
in order to compare them.
SSAP, Dali, Foldminer, Lock, and Geometric Hashing [link] are algorithms which have been
designed in part to align whole protein structures. Despite differences in algorithmic approach, all
of these algorithms essentially evolved from the need to assign the best possible correlation
between points in one structure and points in another. The problem of finding the optimal
alignment is polynomial in the number of atoms in biological data, where we are assured that
atoms cannot fall within a certain distance to each other (Van der Waals forces enforce this), but
without this constraint the problem is exponential.
Protein alignment has been used for the classification and comparison of proteins in many
existing algorithms. These include:
Dali is a structural comparison algorithm based on pairwise distance matrices. Dali uses patterns of residue contacts, similar to contact maps described above in the intramolecular
distances section, in order to align structures. The alignments are found using a randomized
(Monte Carlo) search.
FoldMiner and LOCK 2. FoldMiner finds protein structures similar to an input structure by performing alignment the query structures secondary structure elements with proteins in its
database using the LOCK 2 algorithm. LOCK 2 uses a combination of geometric hashing [link]
and dynamic programming to optimize the alignments of secondary structure elements of
different proteins. Once a set of alignments to similar structures are found, motifs consisting of
similar secondary structure arrangements are constructed and used to refine the similarity
search.
Sequential Structure Alignment Program (SSAP) Given two protein structures, SSAP returns a structural alignment.
Protein Classification
Once alignment algorithms have been implemented, it is possible to explore different
classifications of proteins. Naturally, it would be intuitive to classify proteins solely according to
their structure, but much richer data is available as well. Current classifications of proteins
integrate sequence and structure information in order to maximize their utility. These include the
following:
SCOP (Structural Classification of Protein) is a database of all proteins whose structures have been determined, organized by family (evolutionary relationship), superfamily (structural
and functional similarity), and fold (similar secondary structure, with similar arrangement and
topological connections). SCOP was constructed largely by manual inspection and annotation.
CATH Protein Structure Classification is another database of protein structures, organized
by Class (secondary structure content), Architecture (orientation of secondary structures),
Topology (overall shape and connectivity), and Homologous Superfamily (evolutionary
relationship inferred based on sequence and structure similarity). CATH uses SSAP (the
Sequential Structural Alignment Program, a secondary structure element-based method) for
structural comparison.
The biological purpose of designing Global Stuructural Alignment algorithms was the
identification, classification, and ultimately prediction, of protein function, under the hypothesis
that protein structure dictates protein function. Simultaneously, it was realized that small changes
to proteins could lead to massive changes in function, or nothing at all. This suggested that global
alignment and global structure comparison (as well as global sequence alignment and global
sequence comparison) should not be the only tools used for function prediction.
In particular, it was realized that active sites, clusterings of amino acids on the surface of proteins
and a tiny minority of the entire protein, were often strongly related to protein function. In a
continuation of the effort to predict protein function through structural comparison, algorithms
were developed to compare functionally relevant substructures of proteins. We refer to these
algorithms collectively as local structure alignment algorithms.
Local Matching: Geometric Hashing, Pose Clustering and Match
Augmentation
Algorithms for local structure alignment address the similar computational problem of selecting a
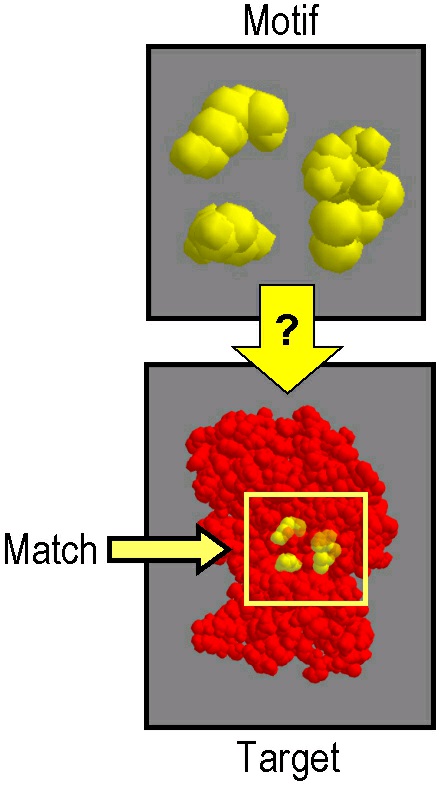
correspondence between a motif, a tiny substructure of a protein, often between 3 and 20 amino
acids, and a target, a full protein structure. Once a correspondence has been established, the
"distance" of the motif to the identified part of the target is measured using lRMSD.
Figure 67.
Some algorithms for local structure alignment are based on pattern matching algorithms. Pattern
matching algorithms seek target substructures called matches which have maximal geometric
similarity to the motif. An excellent example of this type of algorithm is Geometric Hashing
[link], a very flexible framework for geometric pattern matching under noisy constraints.
Geometric Hashing [link], has been adapted to alignment by atom position [link], by backbone C-alpha [link], multiple structural alignment [link], and alignment of hinge-bending and flexible protein models [link]. Other algorithms for substructural alignment include JESS [link], PINTS
[link], webFEATURE [link], and pvSOAR [link]. The description below concentrates on the work developed in [link].
Motifs
A motif S is a set of m points s 1, ..., s m in three dimensions, whose coordinates are taken from
backbone and side-chain atoms. Each motif point s i in the motif has an associated rank p( s i) , a measure of the functional significance of the motif point. Each s i also has a set of alternate amino
acid labels l( s i) in {GLY, ALA, ...}, which represent residues this amino acid has mutated to
during evolution. Labels permit our motifs to simultaneously represent many homologous active
sites with slight mutations, not just a single active site. In this paper, we obtain labels and ranks
using Evolutionary Trace (ET) [link], [link].
Other motifs have been designed with other approaches. Motifs have been composed of points on
the Connolly surface [link] representing electrostatic potentials [link], of hinge-bending sets of points in space [link], of sets of "pseudo-centers" representing protein-ligand interactions [link], or of points taken from atom coordinates with evolutionary data [link], [link], to name a few.
Depending on how motif points are defined, they have different labels associated with them and
these labels need to be taken into account when comparing motifs.
Protein Function Prediction
Local structure comparison algorithms mainly target the biological problem of Protein Function
Prediction. Ideally, a function prediction pipeline should address the following subproblems:
The design of effective motifs
The efficient identification of matches
The effective filtering of matches
Suppose for a moment that we have hand-designed motifs. In the next section, we concentrate on
an efficient algorithm for local structure comparison.

Identification of Matches
Many such algorithms exist, but differ fundamentally in that they are optimized for comparing
different types of motifs. There are algorithms for comparing graph-based motifs [link],
algorithms for finding catalytic sites [link], and the seminal Geometric Hashing framework [link]
which can search for many types of motifs, including motifs based on atom position [link], points on Connolly face centers [link], catalytic triads [link], and flexible protein models [link]. Match Augmentation (MA) [link] is described below.
Match Augmentation
Rank information prioritizes motif data and MA was designed in a prioritized fashion, where
correspondences with higher ranked points are identified first. MA is composed of two parts: Seed
Matching and Augmentation. The purpose of Seed Matching is to identify a match for the seed S'
= { s 1, s 2, s 3}, the three highest ranked motif points. The k lowest lRMSD seed matches are passed to Augmentation to be iteratively expanded into matches for the remaining motif points, in
descending rank order. Augmentation outputs the match with smallest lRMSD.
Seed Matching
Seed Matching identifies all sets of 3 target points T' = { t A, t B, t C} which fulfill our matching criteria with the highest ranked 3 motif points, S'= { s 1, s 2, s 3 }. In this stage, we represent the target as a geometric graph with colored edges. There are exactly three unordered pairs of points in S',
and we name them red, blue and green. In the target, if any pair of target points t i, t j fulfills our
first two criteria with either red, blue or green, we draw a corresponding red blue or green edge
between t i, t j in the target. Once we have processed all pairs of target points, we find all three-
colored triangles in T. These are the Seed Matches, a set of three-point correlations to S' that we
sort by lRMSD and pass to Augmentation.
Augmentation
Augmentation is an application of depth first search that begins with the list of seed matches.
Assuming that there are more than four motif points, we must find correspondences for the
unmatched motif points within the target. Interpret the list of seed matches as a stack of partially
complete matches. Pop off the first match, and considering the lRMSD alignment of this match,
plot the position p of the next unmatched motif point s i relative to the aligned orientation of the
motif. In the spherical region V around p, identify all target points t i, compatible with s i, inside V.
Now compute the lRMSD alignment of all correlated points, include the new correlation ( s i, t i). If
the new alignment satisfies our first two criteria and there are no more unmatched motif points,
put this match into a heap which maintains the match with smallest lRMSD. If there are more
unmatched motif points, put this partial match back onto the stack. Continue to test correlations in
this manner, until V contains no more target points that satisfy our criteria. Then, return to the
stack, and begin again by popping off the first match on the stack, repeating this process until the
stack is empty.
Filtering Matches
Structural similarity is important to functional annotation only if a strong correlation exists
between identifiably significant structural similarity and functional similarity. However, the
existence of a match alone does not guarantee functional similarity. lRMSD can be a
differentiating factor. If matches of homologous proteins represent statistically significant
structural similarity over what is expected by random chance, we could differentiate on lRMSD,
as long as we can evaluate the statistical significance of the lRMSD of a match.
BLAST [link] first calculated the statistical significance of sequence matches with a
combinatorial model of the space of similar sequences. Determining the statistical significance of
structural matches has also been attempted. Modeling was applied for the PINTS database [link]
to estimate the probability of a structural match given a particular LRMSD. An artificial
distribution was parameterized by motif size and amino acid composition in order to fit a given
data set, and the p-value is calculated relative to that distribution. Another approach was taken in
the algorithm JESS [link], using comparative analysis to generate a significance score relative to a specific population of known motifs. Both methods have some disadvantages. The artificial
models of PINTS are not parameterized by the geometry of motifs, and, all else equal, produce
identical distributions for motifs of different geometry. JESS, on the other hand, is dependent on a
set of known motifs; should this set change, all significance scores would have to be revised.
Local structural alignment methods operate on the assumption that local structural and chemical

similarity implies functional similarity. A statistical model has been developed that can be used to
identify the degree of similarity sufficient to follow this implication. Given a match m with
lRMSD r between motif S and target T, exactly one of two hypotheses must hold:
H 0: S and T are structurally dissimilar
H A: S and T are structurally similar
The proposed statistical model implements this measurement by computing a motif profile. Motif
profiles are frequency distributions (see Figure below) of match lRMSDs between S and the entire
Protein Data Bank (PDB) [site], which is essentially a large set of functionally unrelated proteins.
A motif profile is essentially a histogram, where the vertical axis measures the number of matches
at each specific lRMSD, which is measured on the horizontal axis. Motif profiles provide very
complete information about matches typical of H 0. If we suspect that a match m has lRMSD r
indicative of functional similarity, we can use the motif profile to determine the probability p of
observing another match m' with smaller lRMSD by computing the volume under the curve to the
left of r, relative to the entire volume. The probability p, often referred to as the p-value, is the
measure of statistical significance. With a standard of statistical significance alpha, if p is less
than alpha, then we say that the probability of observing a match m' with lRMSD r' less than r is
so low that we reject the null hypothesis in favor of the alternative hypothesis. This process means
that if a match m with lRMSD r has a p-value exactly equal to alpha, then this lRMSD is the
highest lRMSD for which our statistical model predicts that m identifies structural and chemical
similarity sufficient to imply functional similarity. Matches with this property are considered to
be statistically significant.
Figure 68.
The use of motif profiles.
Designing Effective Motifs
Given a motif representing a specific biochemical function, the task of any algorithm seeking to
predict protein functions is to identify other proteins with similar biological functions. This is
immediately affected by how well the motif represents the biological function - protein structures
are flexible, dynamic objects, and any computational representation of these objects is likely to be
inaccurate in some manner. In particular, since it is the computational representation that is
compared, the fidelity of representation is essential for the effectiveness of the structural
comparison approach.
An effective motif must simultaneously fulfill two criteria:
The motif must be sensitive
The motif must be specific
Motifs must maintain geometric and chemical similarity, in respect to the characteristics
compared by the comparison algorithm, to functional homologs. This ensures that algorithms
searching for the motif identify proteins with similar function.
Sensitivity is measured as the proportion of acceptable matches to functional homologs relative to
the total number of functional homologs. Specific motifs must maintain geometric and chemical
dissimilarity, in respect to the characteristics compared by the comparison algorithm, to
functionally unrelated proteins. This ensures that algorithms searching for the motif accidentally
identify less matches to functionally unrelated proteins. Specificity is measured as the proportion
of rejected matches to functionally unrelated proteins, relative to all functionally unrelated
proteins. The ideally effective motif is 100% sensitive and 100% specific.
Under most conditions, effective motifs have been designed by hand. However, a few recently
studies exist which examine the relationship between elements of the motif and the sensitivity and
specificity of the motif. These studies resulted in the design of MultiBind [link], an algorithm which identifies conserved binding patterns among functionally homologous active sites, in order
to generate motifs which represent only conserved binding patterns. Another study of motif design
produced Geometric Sieving [link], an algorithm for maximizing the Geometric Uniqueness of motifs from functionally unrelated proteins.
Putting together effective motif design, an efficient matching algorithm, and a statistical scoring
of the results can lead to an automated functional annotation pipeline for proteins.
Recommended Reading:
Chen, B.Y. (2005). Algorithms for Structural Comparison and Statistical Analysis of 3D
Protein Motifs. Proceedings of the Pacific Symposium on Biocomputing 2005, pp. 334-345.
Chen, B.Y. (2006). Geometric Sieving: Automated Distributed Optimization of 3D Motifs
for Protein Function Prediction. Proceedings of the Tenth Annual Conference on Research in
Computational Molecular Biology (RECOMB 2006), pp. 500-512.
Yao, H., Kristensen, D., Mihalek, I., Sowa, M., Shaw, Ch., Kimmel, M., Kavraki, L. E., and
Yao, H., Kristensen, D., Mihalek, I., Sowa, M., Shaw, Ch., Kimmel, M., Kavraki, L. E., and
Lichtarge, O. (2003). An Accurate, Sensitive, and Scalable Method to Identify Functional
Sites in Protein Structures. Journal of Molecular Biology, 325:255-261.
David M. Kristensen, Brian Y. Chen, Viacheslav Y. Fofanov, R. Matthew Ward, A. Martin